Volume 11, Number 6—June 2005
Research
Global Spread of Vancomycin-resistant Enterococcus faecium from Distinct Nosocomial Genetic Complex
Abstract
Vancomycin-resistant enterococci (VRE) have caused hospital outbreaks worldwide, and the vancomycin-resistance gene (vanA) has crossed genus boundaries to methicillin-resistant Staphylococcus aureus. Spread of VRE, therefore, represents an immediate threat for patient care and creates a reservoir of mobile resistance genes for other, more virulent pathogens. Evolutionary genetics, population structure, and geographic distribution of 411 VRE and vancomycin-susceptible Enterococcus faecium isolates, recovered from human and nonhuman sources and community and hospital reservoirs in 5 continents, identified a genetic lineage of E. faecium (complex-17) that has spread globally. This lineage is characterized by 1) ampicillin resistance, 2) a pathogenicity island, and 3) an association with hospital outbreaks. Complex-17 is an example of cumulative evolutionary processes that improved the relative fitness of bacteria in hospital environments. Preventing further spread of this epidemic E. faecium subpopulation is critical, and efforts should focus on the early disclosure of ampicillin-resistant complex-17 strains.
The emergence of vancomycin-resistant enterococci (VRE) followed a worst-case scenario for nosocomial pathogens: the first VRE isolates that harbored the vanA transposon were identified in 1987 in Europe (1,2), and within 10 years VRE represented >25% of enterococci associated with bloodstream infections in hospitalized patients in the United States (3).
Enterococci are normal inhabitants of the gastrointestinal tract of humans and animals. Two species cause most enterococcal infections, Enterococcus faecalis and E. faecium. The relative importance of E. faecium as a pathogen has increased with the occurrence of high-level resistance to multiple antimicrobial drugs, such as ampicillin and vancomycin (4). The rapid increase of vancomycin resistance compromises physicians' ability to treat infections caused by many of these strains because often no other antimicrobial drugs are available. The epidemiology of VRE infection differs between Europe and the United States. In Europe, VRE are frequently isolated from farm animals, which have been associated with the abundant use of avoparcin as a growth promoter in the agricultural industry, until it was banned in 1997 (5). The reported prevalence of VRE in hospitals has been low, but increasing rates (>10%) in stool and clinical samples were reported recently (6–9). In the United States, avoparcin was never approved for use in agriculture, and neither were any other glycopeptides; consequently, VRE have not been found in animals or healthy persons. However, nosocomial VRE infection and transmission have occurred much more frequently in the United States. Recent reports have documented, in hospitalized patients, horizontal transfer of the vanA gene from vancomycin-resistant E. faecalis to methicillin-resistant Staphylococcus aureus (MRSA), creating MRSA with high-level resistance to vancomycin (10–13). Nosocomial spread of VRE may therefore create a reservoir of mobile resistance genes for other, more virulent, nosocomial pathogens. Without extensive control measures, large-scale emergence of vancomycin-resistant S. aureus (VRSA) may be the next stage in the global crisis of antimicrobial resistance.
The existence of VRE in different ecologic niches complicates the understanding of its epidemiology. Although previous molecular epidemiologic studies on limited numbers of strains suggested host specificity and overrepresentation of certain clones in hospital outbreaks (14,15), these studies did not elucidate the patterns of evolutionary descent among VRE. We determined the population structure of 411 VRE and vancomycin-susceptible E. faecium (VSE) isolates by using multilocus sequence typing (MLST), explored the evolutionary origin of epidemic isolates associated with documented hospital outbreaks and other isolates, and assessed the association with ampicillin resistance and the presence of a recently discovered putative pathogenicity island (PAI) in E. faecium (16).
The strain collection included 5 categories of VRE and VSE: 1) 96 animal surveillance (bison, calves, cats, dogs, ostriches, poultry, pigs, rodents) isolates (43 VRE, 53 VSE) from 7 countries in Africa and Europe; 2) 57 epidemiologically unrelated community surveillance isolates (20 VRE, 37 VSE) from nonhospitalized persons from 7 countries in Australia and Europe; 3) 64 epidemiologically unrelated surveillance (fecal) isolates (45 VRE, 19 VSE) from hospitalized patients not linked to hospital outbreaks from 9 countries in Australia, Europe, and North and South America; 4) 162 epidemiologically unrelated hospital isolates (43 VRE, 118 VSE, 1 not determined) from clinical specimens (blood, pus, and urine) from 17 countries in Africa, Australia, Europe, and North and South America; and 5) 1 strain from each of 32 different documented hospital outbreaks (28 VRE, 4 VSE) in 10 countries in Australia, Europe, and North and South America (W. Grubb and D. Jonas, pers. comm.; 15,17–23).
We determined vancomycin susceptibilities for 410 isolates and ampicillin susceptibilities for 381 isolates by using standard agar dilution methods according to NCCLS guidelines. Isolates with MIC ≥16 μg/mL for ampicillin and ≥8 μg/mL for vancomycin were considered to be resistant. In total, 394 strains were screened for the esp gene with primer sets and amplification conditions described previously (24). Independent and combined effects of virulence and resistance markers on the abundance of complex-17 were estimated by using multiple logistic regression analysis (Stata 7.0, StataCorp LP, College Station, TX, USA).
MLST was carried out with a standard set of primers that amplify the 7 genes included in the E. faecium MLST scheme (14). Information on these loci, the latest set of primers, amplification conditions, and details of all isolates are available on the MLST Web site (http://efaecium.mlst.net).
The eBURST program was used to assess the genetic relationships of genotypes, to assign isolates to genetic complexes, and to study patterns of evolutionary descent of isolates within a complex (25). Complexes were identified by using the stringent (6/7 shared alleles) group definition with 1,000 bootstrap replicates. The BLAND program was used to examine the relationship between pairwise allelic differences and nucleotide sequence differences (26). If genetic diversity in E. faecium is mainly the result of accumulated point mutations, then recently diverged strains will have a high level of similarity in both their allelic profiles as well as in the nucleotide sequence of the nonidentical alleles, which results in a positive correlation between the number of nucleotide differences in nonidentical alleles and the number of allelic differences. However, such a trend will be absent when recombination plays an important role in generating the genetic diversity, since nonidentical alleles of closely related isolates can differ at multiple nucleotide sites.
To assess the effect of recombination on the population structure of E. faecium in more detail, the topologies of the 7 MLST gene trees were compared by using the Shimodaira-Hasegawa test (27). Briefly, maximum likelihood trees for each MLST gene were obtained under a general time-reversible model, with a proportion of invariant sites and rate heterogeneity among sites assuming a discrete gamma distribution with 8 categories (GTR+I+Γ model). PAUP* 4.0b10 was used to obtain the maximum likelihood trees by using a neighbor-joining starting tree followed by tree-bisection reconnection branch swapping (28). For a given gene, the Shimodaira-Hasegawa test compares the difference in log likelihoods of competing tree topologies. A null distribution of differences in log likelihoods was obtained by 1,000 replicates of nonparametric bootstrapping of reestimated log likelihoods. We conducted 107 Shimodaira-Hasegawa tests for each MLST gene by comparing the 7 MLST gene trees and 100 random trees separately generated for each of the MLST genes. In a clonal population, the different MLST housekeeping genes have similar tree topologies, but with recombination, the different genes may have different tree topologies that may fit random trees better.
Associations between ampicillin resistance, presence of a novel putative E. faecium PAI, and genetic clustering in complex-17 were described by linear logistic regression models: log odds = b0 + b1x1 + b2x2 + b3x3 + … + bixi. Log odds denotes the natural logarithm of the proportion of samples from an epidemiologic group belonging to complex-17, bi denotes the parameter estimated by maximum likelihood methods and xi the level of exposure, e.g., 0 and 1 for ampicillin resistance, vancomycin resistance, and the presence of PAI and 0–4 for the epidemiologic source of isolates: animal surveillance, community surveillance, hospital surveillance, clinical sample, and hospital outbreak, respectively.
Identification of Clonal Lineages
Figure 1
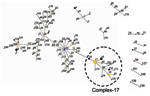
Figure 1. . Clustering of 175 sequence types representing 411 isolates with eBURST (25). This algorithm identifies the founder of a complex or genogroup of related sequence types (ST) and subsequent patterns of...
MLST of 411 E. faecium isolates resulted in 175 different sequence types (ST). Clustering these types with the eBURST algorithm (25) showed 1 large complex of genetically related types. ST-22 was the primary founder; 3 minor complexes had ST-1, -69, and -94 as primary founders; 6 complexes had only 2 or 3 STs; and 57 singletons were not linked to the aforementioned complexes (Figure 1). Within complex-22, ST-17 represents an important secondary founder of a distinct branch designated complex-17.
Selective Advantage of the Successful Hospital-adapted Complex-17
In all, 142 of 411 isolates belonged to complex-17, with a gradual increase in proportion among animal isolates (1/96), human community isolates (3/57), human hospital surveillance isolates (15/64), and human clinical isolates (95/162), to hospital-outbreak isolates (28/32) (Table 1). Ampicillin resistance, presence of the E. faecium PAI (16), and genetic clustering in complex-17 were strongly associated (Table 2).
Figure 2
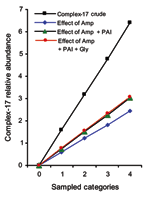
Figure 2. . Relative abundance of complex-17 in various sampled categories and proportion increase explained by combined effect of 3 parameters. 0, animal surveillance samples; 1, human community surveillance samples; 2, human hospitalized...
When controlling for individual and combined effects of ampicillin resistance, presence of PAI, and vancomycin resistance, we can show that 1) the loglinear assumption holds for all effect parameters, and linear models describe the observed frequencies without substantial loss of goodness of fit; 2) individual genetic markers exert an independent and multiplicative effect; and 3) all genetic markers combined explain ≈48% of the category-specific abundance of complex-17 (Figure 2). The effect of vancomycin resistance did not increase the explanatory value of the model, owing to the fact that determinants for vancomycin resistance could be found in equal proportions within and outside of complex-17, likely a result of widespread horizontal transfer of vanA (Table 1). This finding suggests that the epidemiologic success of descendants of ST-17 that results in clinical infections and hospital epidemics was at least partly related to antimicrobial resistance and the presence of putative virulence genes. The fact that 126 of 128 isolates of complex-17 were resistant to ampicillin and only 77 of 139 isolates of complex-17 contain PAI (Table 1) suggests that E. faecium acquired ampicillin resistance first, which resulted in a selective advantage in hospitals, followed by the acquisition of PAI, which further facilitated transmission.
Estimates of Recombination
Figure 3
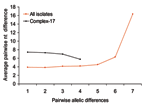
Figure 3. . Sequence diversity versus allelic diversity. The average number of nucleotide (nt) differences in nonidentical alleles for all pairwise comparisons of the 178 Enterococcus faecium sequence types (STs), and the 15...
To assess the effect of recombination on the population structure of E. faecium, we estimated whether single locus variants (SLVs) from the presumed founders of complex-1, -17, -22, -69, and -94 have arisen by point mutations or by recombination (Table 3). Of all allelic differences between ancestor-SLV pairs (n = 30), 22 (4 in complex-17) included >1 nucleotide, were found in multiple clonal complexes, and thus were most likely a result of recombination. Eight allelic differences (2 in complex-17) included only a single nucleotide change, were unique within the dataset, and thus were most likely a result of mutation. Therefore, most alleles of SLVs in complex-17 and in the other complexes have arisen by recombination in the initial stages of diversification rather than by de novo point mutation. An important role for recombination in genetic diversification in E. faecium was confirmed by the lack of a positive trend between the number of nucleotide differences in nonidentical alleles and the number of allelic differences (Figure 3) (26). The finding of high average numbers (≥4) of nucleotide differences in the nonidentical alleles of SLVs in the total E. faecium population as well as in complex-17 also points towards frequent recombination.
The degree of phylogenetic congruence between the 7 MLST genes was examined in a set of 24 diverse STs. These 24 STs were separated from each other by a linkage distance of >0.4 on a UPGMA (unweighted pair-group method with arithmetic mean) tree constructed from the pairwise comparisons of their allelic profiles (data not shown) and included the primary founders of CC1 (ST1), CC22 (ST22), CC69 (ST69), and CC94 (ST94); secondary founders of important subgroups complex-5 (ST5) and complex-17 (ST17); 1 ST (ST76) belonging to a small complex of 3 STs; and 17 singletons (STs 15, 38, 39, 54, 67, 74, 83, 84, 89, 96, 98, 99, 101, 107, 118, 142, 163). The results of the congruence analysis presented in Table 4 show that 25 (60%) of 42 of the pairwise comparisons of the 7 MLST loci were incongruent. Of the 7 genes, atpA is the most incongruent. This analysis confirms that recombination played a substantial role in the evolution of E. faecium.
Nosocomial VRE, which rapidly emerged in the United States in the 1990s after their initial discovery in Europe, are found in increasing rates in hospitals in Europe, Asia, and South America (5–7,9,23,29,30). The data presented in this study show that most of these hospital-derived VRE are part of a single clonal lineage. This lineage, designated complex-17 after its presumed founder ST-17, represents most hospital outbreak and clinical isolates, apparently because it successfully adapted to hospital environments.
The >400 strains analyzed in this study were selected from a large representative collection of 2,000 E. faecium isolates. A wide variety of sources were used as selection criteria: hospital-associated outbreaks; clinical samples and stool samples from hospitalized patients, healthy persons, and animals; and a wide geographic distribution (21 countries on 5 continents).
Complex-17 probably evolved from the primary E. faecium ancestor ST-22 through a combination of mutation and recombination. The following observations suggest that recombination has been especially important in the genetic diversification of the E. faecium population: 1) within clonal complexes, most SLVs (73%) have arisen by recombination rather than point mutations; 2) no positive correlation exists between the degree of allelic diversity and the number of nucleotide differences in nonidentical alleles; and 3) most (60%) of the comparisons of MLST gene tree topologies were incongruent.
Figure 4
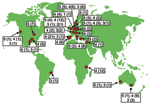
Figure 4. . Global distribution of complex-17 isolates. Red circles indicate cities where complex-17 isolates were recovered. Numbers indicate epidemiologic sources: 1, animal isolates; 2, human community surveillance isolates; 3, surveillance (feces) isolates...
Exploitation of a novel ecologic niche as hospital settings by E. faecium ST-17 often starts with adaptive changes (31). On the basis of our findings, we postulate that ST-17 acquired ampicillin resistance and a novel putative PAI. This amplifying selective process in which variants with a selective advantage can more easily acquire additional adaptive mechanisms has been called "genetic capitalism" (32). After successfully exploiting the hospital environment, ST-17 increased in frequency to become the dominant clone. Genetic diversification over time finally resulted in a meroclone, complex-17, of highly related genotypes, fully adapted as a nosocomial pathogen, that has spread globally (Figure 4). In addition, E. faecium STs, predominantly ST-78, belonging to complex-17, have recently also been found in the Republic of South Korea (K.S. Ko and J.-H. Song, pers. comm.). Considering the short period in which multiresistant E. faecium emerged as a nosocomial pathogen (33), complex-17 represents the first globally dispersed nosocomial-adapted clonal lineage. Despite the frequency of recombination events, clonal complex-17 is still detectable within the E. faecium population, which suggests that the emergence of this complex is relatively recent.
The existence of epidemic clones, even in recombining populations, is also seen in other bacterial species (34,35). However, the evolution of a single epidemic and clinically relevant genetic complex, as seen with E. faecium, differs from the evolution of other gram-positive pathogens like Streptococcus pneumoniae and Staphylococcus aureus. In S. pneumoniae, pandemic clones such as ST81, ST90, and ST156 represent major invasive and multidrug-resistant isolates that have spread globally (36). The allelic profiles of these clones, however, are highly diverse, which suggests that they are genetically unrelated and do not constitute a single genetic lineage, as does E. faecium. Furthermore, the serotype of S. pneumoniae seems a more important marker of invasiveness than the overall genotype (37). In S. aureus isolates, major pandemic MRSA clones that are responsible for most hospital-acquired infections are found in multiple genetically unrelated lineages, though most previously identified pandemic clones are found in clonal complex 8 (38,39). Therefore, the genetic diversity of major epidemic clones as seen in S. pneumoniae or S. aureus may not have yet emerged in E. faecium epidemic populations.
Stress-inducing conditions in hospitals, such as antimicrobial drug use, may have favored the selection of an enterococcal subpopulation, complex-17, with enhanced antibacterial resistance, virulence, and ability to spread. Whether reducing antimicrobial selection pressure in hospitals will reestablish a susceptible and less transmissible enterococcal population is unknown and will at least partly depend on the relative fitness costs of sustaining antimicrobial resistance and virulence determinants in E. faecium. Furthermore, the hospital-adapted complex-17 has rapidly spread globally during the last 2 decades. Subsequent acquisition of vanA- or vanB- containing transposons by horizontal gene transfer resulted in VRE with pandemic potential. Rapid diagnosis of complex-17 strains based on multiple locus variable number of tandem repeat analysis (MLVA) may help control its spread (40). Whether this effort will be successful depends on the level of complex-17 endemicity in the hospital. In many European countries, a relatively large community reservoir of VRE exists, a result of the massive use of the antimicrobial drug avoparcin as a growth promoter, while in general the prevalence of hospital-adapted (complex-17) VRE is much lower. In such a setting, hospital transmission of isolates belonging to complex-17 can be halted by using a fast genotyping scheme like MLVA to discriminate between hospital-adapted (complex-17) and community E. faecium strains followed by strict infection control measures. The combination of infection control measures plus genotyping controlled an outbreak of VRE in a Dutch hospital (41).
Establishing nosocomial co-endemicity of VRE and MRSA will facilitate the horizontal transfer of vanA- or vanB-containing transposons, transforming MRSA into VRSA, with implications for patient care. Until now, 3 sporadic cases of vanA-induced VRSA have been reported in the United States in 2002 and 2004. Spread of multidrug-resistant E. faecium strains and their resistance genes will have serious implications for health care, and control efforts should focus on early detection of E. faecium isolates belonging to complex-17.
Dr. Willems began this research at the National Institute for Public Heath and the Environment and continued at the University Medical Center Utrecht, where he is currently working. His research interests are the molecular epidemiology, population structure, and genetic evolution of multidrug-resistant nosocomial pathogens.
Acknowledgments
We thank B. Blomberg, A. Sundsfjord, A. van Belkum, D. Tribe, E. de Leener, W. Grubb, D. Jonas, M.G. Bonora, R.C. Zanella, L. Baldassarri, N. Shankar, N. Woodford, M. Soltani, S.C. Mohn, R. Jureen, and K. Borgen for providing E. faecium strains; N. Shankar, M. Gilmore, J. van Embden, B. Levin, and W. Jansen for critical reading of the manuscript; and D. Aanensen and B. Spratt for hosting the E. faecium MLST database (www.mlst.net) at Imperial College London.
Funding for this study was provided by University Medical Center Utrecht and the National Institute for Public Health and the Environment, the Netherlands. The funding sources had no role in the selection, design, or reporting of this study.
References
- Leclercq R, Derlot E, Duval J, Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med. 1988;319:157–61. DOIPubMedGoogle Scholar
- Uttley AH, Collins CH, Naidoo J, George RC. Vancomycin-resistant enterococci. Lancet. 1988;1:57–8. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. National nosocomial infections surveillance (NNIS) system report, data summary from January 1992–June 2001, issued August 2001. Am J Infect Control. 2001;29:404–21. DOIPubMedGoogle Scholar
- Iwen PC, Kelly DM, Linder J, Hinrichs SH, Dominguez EA, Rupp ME, Change in prevalence and antibiotic resistance of Enterococcus species isolated from blood cultures over an 8-year period. Antimicrob Agents Chemother. 1997;41:494–5.PubMedGoogle Scholar
- Bonten MJ, Willems R, Weinstein RA. Vancomycin-resistant enterococci: why are they here, and where do they come from? Lancet Infect Dis. 2001;1:314–25. DOIPubMedGoogle Scholar
- Bouchillon SK, Johnson BM, Hoban DJ, Johnson JL, Dowzicky MJ, Wu DH, Determining incidence of extended spectrum beta-lactamase producing Enterobacteriaceae, vancomycin-resistant Enterococcus faecium and methicillin-resistant Staphylococcus aureus in 38 centres from 17 countries: the PEARLS study 2001–2002. Int J Antimicrob Agents. 2004;24:119–24. DOIPubMedGoogle Scholar
- European Antimicrobial Resistance Surveillance System (EARSS). EARSS annual report 2003. [cited 2005 Jan 17]. Available from http://www.earss.rivm.nl/PAGINA/DOC/EARSS%20annual%20report.2003.pdf
- Goossens H, Jabes D, Rossi R, Lammens C, Privitera G, Courvalin P. European survey of vancomycin-resistant enterococci in at-risk hospital wards and in vitro susceptibility testing of ramoplanin against these isolates. J Antimicrob Chemother. 2003;51(Suppl 3):iii5–12. DOIPubMedGoogle Scholar
- Jones ME, Draghi DC, Thornsberry C, Karlowsky JA, Sahm DF, Wenzel RP. Emerging resistance among bacterial pathogens in the intensive care unit—a European and North American Surveillance study (2000–2002). Ann Clin Microbiol Antimicrob. 2004;3:14. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. Vancomycin-resistant Staphylococcus aureus—New York, 2004. MMWR Morb Mortal Wkly Rep. 2004;53:322–3.PubMedGoogle Scholar
- Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, Downes FP, Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N Engl J Med. 2003;348:1342–7. DOIPubMedGoogle Scholar
- Tenover FC, Weigel LM, Appelbaum PC, McDougal LK, Chaitram J, McAllister S, Vancomycin-resistant Staphylococcus aureus isolate from a patient in Pennsylvania. Antimicrob Agents Chemother. 2004;48:275–80. DOIPubMedGoogle Scholar
- Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science. 2003;302:1569–71. DOIPubMedGoogle Scholar
- Homan WL, Tribe D, Poznanski S, Li M, Hogg G, Spalburg E, Multilocus sequence typing scheme for Enterococcus faecium. J Clin Microbiol. 2002;40:1963–71. DOIPubMedGoogle Scholar
- Willems RJL, Top J, van den Braak N, van Belkum A, Endtz H, Mevius D, Host specificity of vancomycin-resistant Enterococcus faecium. J Infect Dis. 2000;182:816–23. DOIPubMedGoogle Scholar
- Leavis H, Top J, Shankar N, Borgen K, Bonten M, van Embden J, A novel putative enterococcal pathogenicity island linked to the esp virulence gene of Enterococcus faecium and associated with epidemicity. J Bacteriol. 2004;186:672–82. DOIPubMedGoogle Scholar
- Bonora MG, Bordin C, Bragagnolo L, Cirelli L, de Fatima M, Grossato A, Molecular analysis of vanA enterococci isolated from humans and animals in northeastern Italy. Microb Drug Resist. 2001;7:247–56. DOIPubMedGoogle Scholar
- Bonora MG, Ligozzi M, de Fatima M, Bragagnolo L, Goglio A, Guazzotti GC, Vancomycin-resistant Enterococcus faecium isolates causing hospital outbreaks in northern Italy belong to the multilocus sequence typing C1 lineage. Microb Drug Resist. 2004;10:114–23. DOIPubMedGoogle Scholar
- Jureen R, Top J, Mohn SC, Harthug S, Langeland N, Willems RJL. Molecular characterization of ampicillin-resistant Enterococcus faecium isolates from hospitalized patients in Norway. J Clin Microbiol. 2003;41:2330–6. DOIPubMedGoogle Scholar
- Routsi C, Platsouka E, Willems RJ, Bonten MJ, Paniara O, Saroglou G, Detection of enterococcal surface protein gene (esp) and amplified fragment length polymorphism typing of glycopeptide-resistant Enterococcus faecium during its emergence in a Greek intensive care unit. J Clin Microbiol. 2003;41:5742–6. DOIPubMedGoogle Scholar
- Simonsen GS, Smabrekke L, Monnet DL, Sorensen TL, Moller JK, Kristinsson KG, Prevalence of resistance to ampicillin, gentamicin and vancomycin in Enterococcus faecalis and Enterococcus faecium isolates from clinical specimens and use of antimicrobials in five Nordic hospitals. J Antimicrob Chemother. 2003;51:323–31. DOIPubMedGoogle Scholar
- Willems RJ, Homan W, Top J, van Santen-Verheuvel M, Tribe D, Manzioros X, Variant esp gene as a marker of a distinct genetic lineage of vancomycin-resistant Enterococcus faecium spreading in hospitals. Lancet. 2001;357:853–5. DOIPubMedGoogle Scholar
- Zanella RC, Brandileone MC, Bokermann S, Almeida SC, Valdetaro F, Vitorio F, Phenotypic and genotypic characterization of VanA enterococcus isolated during the first nosocomial outbreak in Brazil. Microb Drug Resist. 2003;9:283–91. DOIPubMedGoogle Scholar
- Leavis HL, Willems RJ, Top J, Spalburg E, Mascini EM, Fluit AC, Epidemic and nonepidemic multidrug-resistant Enterococcus faecium. Emerg Infect Dis. 2003;9:1108–15.PubMedGoogle Scholar
- Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186:1518–30. DOIPubMedGoogle Scholar
- Feil EJ, Cooper JE, Grundmann H, Robinson DA, Enright MC, Berendt T, How clonal is Staphylococcus aureus? J Bacteriol. 2003;185:3307–16. DOIPubMedGoogle Scholar
- Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–6.
- Swofford DL. PAUP*: phylogenetic analysis using parsimony and other methods, version 4. Sunderland (MA): Sinauer Associates, Inc.; 2000.
- Shin JW, Yong D, Kim MS, Chang KH, Lee K, Kim JM, Sudden increase of vancomycin-resistant enterococcal infections in a Korean tertiary care hospital: possible consequences of increased use of oral vancomycin. J Infect Chemother. 2003;9:62–7. DOIPubMedGoogle Scholar
- Taneja N, Rani P, Emmanuel R, Sharma M. Significance of vancomycin resistant enterococci from urinary specimens at a tertiary care centre in northern India. Indian J Med Res. 2004;119:72–4.PubMedGoogle Scholar
- Martinez JL, Baquero F. Interactions among strategies associated with bacterial infection: pathogenicity, epidemicity, and antibiotic resistance. Clin Microbiol Rev. 2002;15:647–79. DOIPubMedGoogle Scholar
- Baquero F, Coque TM, Canton R. Antibiotics, complexity, and evolution. ASM News. 2003;69:547–52.
- Cetinkaya Y, Falk P, Mayhall CG. Vancomycin-resistant enterococci. Clin Microbiol Rev. 2000;13:686–707. DOIPubMedGoogle Scholar
- Morelli G, Malorny B, Muller K, Seiler A, Wang JF, del Valle J, Clonal descent and microevolution of Neisseria meningitidis during 30 years of epidemic spread. Mol Microbiol. 1997;25:1047–64. DOIPubMedGoogle Scholar
- Robinson DA, Briles DE, Crain MJ, Hollingshead SK. Evolution and virulence of serogroup 6 pneumococci on a global scale. J Bacteriol. 2002;184:6367–75. DOIPubMedGoogle Scholar
- Enright MC, Fenoll A, Griffiths D, Spratt BG. The three major Spanish clones of penicillin-resistant Streptococcus pneumoniae are the most common clones recovered in recent cases of meningitis in Spain. J Clin Microbiol. 1999;37:3210–6.PubMedGoogle Scholar
- Brueggemann AB, Griffiths DT, Meats E, Peto T, Crook DW, Spratt BG. Clonal relationships between invasive and carriage Streptococcus pneumoniae and serotype- and clone-specific differences in invasive disease potential. J Infect Dis. 2003;187:1424–32. DOIPubMedGoogle Scholar
- Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, Spratt BG. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci U S A. 2002;99:7687–92. DOIPubMedGoogle Scholar
- Robinson DA, Enright MC. Evolutionary models of the emergence of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47:3926–34. DOIPubMedGoogle Scholar
- Top J, Schouls LM, Bonten MJ, Willems RJ. Multiple-locus variable-number tandem repeat analysis, a novel typing scheme to study the genetic relatedness and epidemiology of Enterococcus faecium isolates. J Clin Microbiol. 2004;42:4503–11. DOIPubMedGoogle Scholar
- Ridwan B, Mascini E, van der Reijden N, Verhoef J, Bonten M. What action should be taken to prevent spread of vancomycin resistant enterococci in European hospitals? BMJ. 2002;324:666–8. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 11, Number 6—June 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Rob J.L. Willems, Eijkman-Winkler Institute for Microbiology, Infectious Diseases and Inflammation, University Medical Center Utrecht, Heidelberglaan 100, 3584 CX Utrecht, the Netherlands; fax: 31-30-254-1776
Top