Volume 18, Number 12—December 2012
Dispatch
Analysis of Complete Puumala Virus Genome, Finland
Figure 2
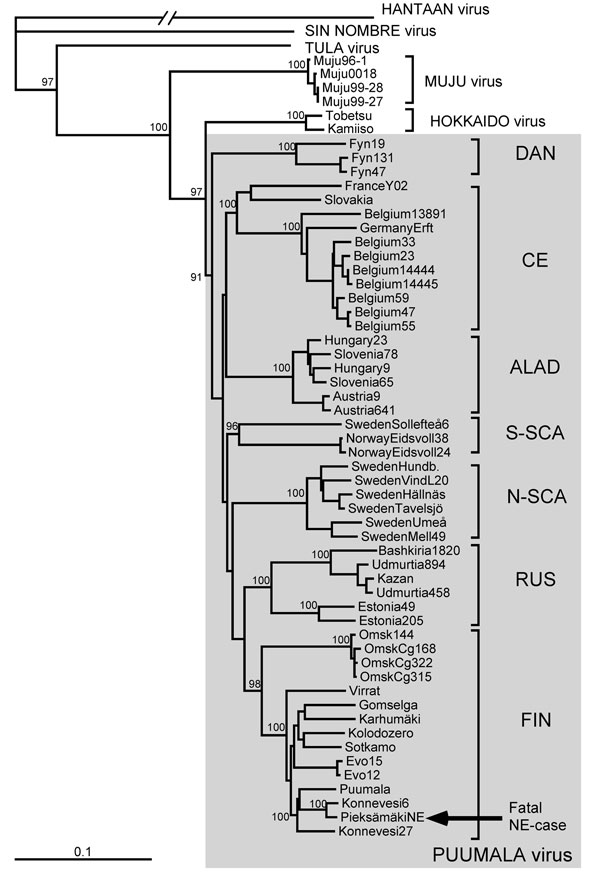
Figure 2. . Phylogenetic tree of PUUV S segment sequences (coding region). Topologies of the M and L trees were similar (not shown). Calculations were performed by using the PHYLIP program package (distributed by J. Felsenstein, University of Washington, Seattle, WA, USA). Five hundred bootstrap replicates were generated by using the SeqBoot program and submitted to the distance matrix algorithm (DNAdist program), with the maximum-likelihood model for nucleotide substitutions). The resulting distance matrices were analyzed by using the neighbor-joining tree-fitting algorithm (Neighbor program). The bootstrap support values were calculated by using the Consense program. Hantavirus sequences used for comparison were recovered from GenBank. Gray shading indicates PUUV strains. PUUV, Puumala virus; S, small; M, medium; L, large; DAN, Danish; CE, Central European; ALAD, Alpe-Adrian; S-SCA, South Scandinavian; N-SCA, North Scandinavian; RUS, Russian; FIN, Finnish; NE, nephropathia epidemica. Scale bar indicates genetic distance of 0.1.