Volume 18, Number 5—May 2012
Dispatch
Origin of Human T-Lymphotropic Virus Type 1 in Rural Côte d’Ivoire
Figure 2
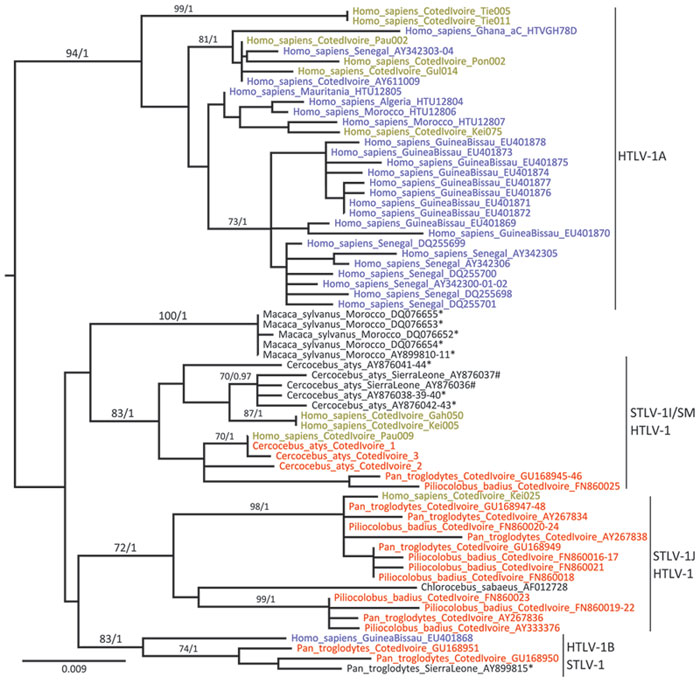
Figure 2. . . Maximum-likelihood tree based on the analysis of a long terminal repeat (853 bp) alignment in a study of the origin of human T-lymphotropic virus type 1 (HTLV-1) in rural western Africa, 2006–2007. Bayesian analyses supported similar topologies. After rooting, branches leading to outgroup sequences (HTVMEL5 and Z46900) were removed from the figure to increase its legibility. The HTLV-1 sequences determined from specimens from West and North Africa are shown in light blue; HTLV-1 sequences determined from persons living in the region of Taï National Park are shown in green; STLV-1 sequences determined from specimens from West and North Africa are shown in black; and STLV-1 sequences determined from persons living in the Taï National Park are shown in red. Sequence names are built as follows: [host species]_[country of origin]_[GenBank accession number]. Reference sequence names also include their molecular subtype assignation: [host species]_[country of origin]_[molecular subtype]_[accession number]. *Sequences determined from captive or semicaptive hosts; #sequences determined from bushmeat samples. Molecular subtypes were assigned on the basis of an analysis performed on an enlarged dataset including assigned reference sequences (data not shown). Bootstrap (Bp) and posterior probability (pp) values are indicated where Bp>50.0 and pp>0.95. Scale bar indicates nucleotide substitutions per site.