Volume 21, Number 10—October 2015
Research
Evolutionary and Ecological Characterization of Mayaro Virus Strains Isolated during an Outbreak, Venezuela, 2010
Albert J. Auguste, Jonathan Liria, Naomi L. Forrester, Dileyvic Giambalvo, Maria Moncada, Kanya C. Long1, Dulce Morón, Nuris de Manzione, Robert B. Tesh, Eric S. Halsey, Tadeusz J. Kochel, Rosa Hernandez, Juan-Carlos Navarro, and Nikos Vasilakis
Figure 2
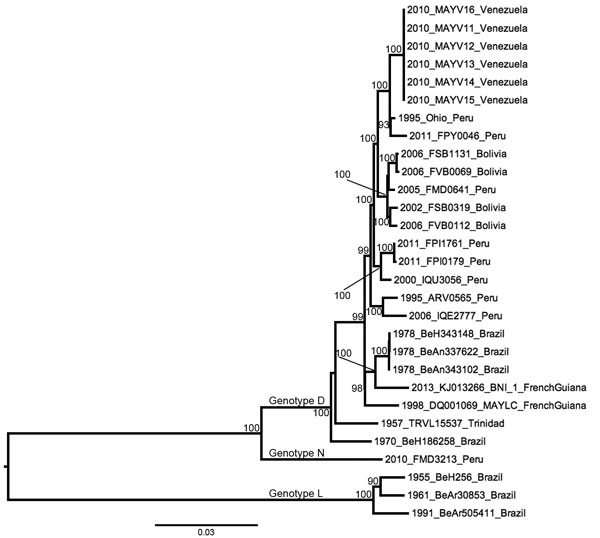
Figure 2. Midpoint-rooted maximum-likelihood phylogeny of 29 Mayaro virus strains on the basis of complete genome sequences, Venezuela, 2010. Nodes are labeled with bootstrap values ≥90%. Tip labels indicate year of isolation, strain name, and country of isolation. Scale bar indicates percentage nucleotide sequence divergence. Isolates DQ001069 and KJ013266 were previously sequenced and obtained from GenBank.
1Current affiliation: Andrews University, Berrien Springs, Michigan, USA.
Page created: September 22, 2015
Page updated: September 22, 2015
Page reviewed: September 22, 2015
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.