Volume 21, Number 12—December 2015
CME ACTIVITY - Dispatch
Life-Threatening Sochi Virus Infections, Russia
Figure 1
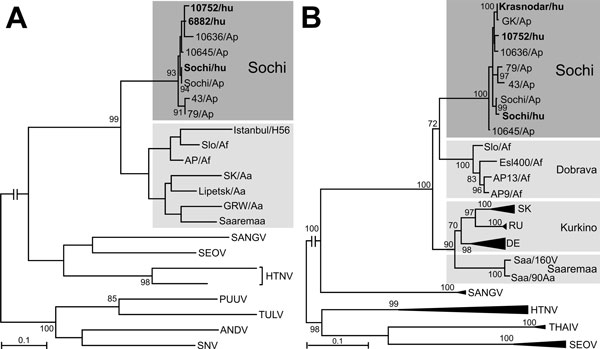
Figure 1. Phylogenetic analysis segment sequences of Sochi virus, Russia: A) 347-bp large (L) segment sequence; B) 1,197-bp small (S) segment sequence. Virus sequences derived from patients (shown in bold type) and Apodemus ponticus mice cluster within the Sochi genotype of DOBV. Evolutionary analysis was conducted in MEGA6 (6). The evolutionary history was inferred by using the maximum-likelihood method based on the Tamura 3-parameter model with a discrete gamma distribution and 5 rate categories (analysis in panel A) and on the general time reversible model with gamma rates and heterogeneous patterns (analysis in panel B), respectively, which were estimated to be the best-fit substitution model according to the Bayesian information criterion. Scale bars indicate an evolutionary distance of 0.1 substitutions per position in the sequence. Bootstrap values >70%, calculated from 500 replicates, are shown at the tree branches. GenBank accession numbers of all sequences used in the analysis are listed in Technical Appendix Table 1). Dark gray shading iindicates cluster of DOBV-Sochi strains; light gray shading indicates different clusters of strains from other DOBV genotypes. ANDV, Andes virus; DOBV, Dobrava-Belgrade virus; HTNV, Hantaan virus; PUUV, Puumala virus; SANGV, Sangassou virus; SEOV, Seoul virus; SNV, Sin Nombre virus; THAIV, Thailand virus; TULV, Tula virus.
References
- Kruger DH, Figueiredo LTM, Song JW, Klempa B. Hantaviruses—globally emerging pathogens. J Clin Virol. 2015;64:128–36. DOIPubMedGoogle Scholar
- Tkachenko EA, Okulova NM, Yunicheva YV, Morzunov SP, Khaĭbulina SF, Riabova TE, The epizootological and virological characteristics of a natural hantavirus infection focus in the subtropic zone of the Krasnodarsk Territory [in Russian]. Vopr Virusol. 2005;50:14–9 .PubMedGoogle Scholar
- Klempa B, Tkachenko EA, Dzagurova TK, Yunicheva YV, Morozov VG, Okulova NM, Hemorrhagic fever with renal syndrome caused by 2 lineages of Dobrava hantavirus, Russia. Emerg Infect Dis. 2008;14:617–25. DOIPubMedGoogle Scholar
- Dzagurova TK, Witkowski PT, Tkachenko EA, Klempa B, Morozov VG, Auste B, Isolation of Sochi virus from a fatal case of hantavirus disease with fulminant clinical course. Clin Infect Dis. 2012;54:e1–4. DOIPubMedGoogle Scholar
- Klempa B, Avsic-Zupanc T, Clement J, Dzagurova TK, Henttonen H, Heyman P, Complex evolution and epidemiology of Dobrava-Belgrade hantavirus: definition of genotypes and their characteristics. Arch Virol. 2013;158:521–9. DOIPubMedGoogle Scholar
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. DOIPubMedGoogle Scholar
- Kramski M, Meisel H, Klempa B, Krüger DH, Pauli G, Nitsche A. Detection and typing of human pathogenic hantaviruses by real-time reverse transcription–PCR and pyrosequencing. Clin Chem. 2007;53:1899–905. DOIPubMedGoogle Scholar
- Okulova NM, Khliap LA, Varshavskii AA, Dzagurova TK, Iunicheva IV, Riabova TE, Spatial structure of natural foci of hantavirus on the territory of northwestern Caucasus [in Russian]. Zh Mikrobiol Epidemiol Immunobiol. 2013; (
Sep–Oct ):47–53 .PubMedGoogle Scholar - Mertz GJ, Hjelle B, Crowley M, Iwamoto G, Tomicic V, Vial PA. Diagnosis and treatment of new world hantavirus infections. Curr Opin Infect Dis. 2006;19:437–42. DOIPubMedGoogle Scholar
- Zhang YZ, Zou Y, Fu ZF, Plyusnin A. Hantavirus infections in humans and animals, China. Emerg Infect Dis. 2010;16:1195–203. DOIPubMedGoogle Scholar
- Noh JY, Cheong HJ, Song JY, Kim WJ, Song KJ, Klein TA, Clinical and molecular epidemiological features of hemorrhagic fever with renal syndrome in Korea over a 10-year period. J Clin Virol. 2013;58:11–7. DOIPubMedGoogle Scholar
- Avsic-Zupanc T, Petrovec M, Furlan P, Kaps R, Elgh F, Lundkvist A. Hemorrhagic fever with renal syndrome in the Dolenjska region of Slovenia—a 10-year survey. Clin Infect Dis. 1999;28:860–5. DOIPubMedGoogle Scholar
- Papa A, Antoniadis A. Hantavirus infections in Greece—an update. Eur J Epidemiol. 2001;17:189–94 . DOIPubMedGoogle Scholar
- Dzagurova TK, Klempa B, Tkachenko EA, Slyusareva GP, Morozov VG, Auste B, Molecular diagnostics of hemorrhagic fever with renal syndrome during a Dobrava virus infection outbreak in the European part of Russia. J Clin Microbiol. 2009;47:4029–36 . DOIPubMedGoogle Scholar