Volume 21, Number 5—May 2015
Research
Transmission of Hepatitis C Virus among Prisoners, Australia, 2005–2012
Abstract
Hepatitis C virus (HCV) is predominantly transmitted between persons who inject drugs. For this population, global prevalence of HCV infection is high and incarceration is common and an independent risk factor for HCV acquisition. To explore HCV transmission dynamics in incarcerated populations, we integrated virus sequences with risk behavior and spatiotemporal data and analyzed transmission clusters among prisoners in Australia. We detected 3 clusters of recent HCV transmission consisting of 4 likely in-custody transmission events involving source/recipient pairs located in the same prison at the same time. Of these 4 events, 3 were associated with drug injecting and equipment sharing. Despite a large population of prisoners with chronic HCV, recent transmission events were identified in the prison setting. This ongoing HCV transmission among high-risk prisoners argues for expansion of prevention programs to reduce HCV transmission in prisons.
Hepatitis C virus (HCV) is a blood-borne virus that infects 3–4 million persons each year (1). In industrialized countries, transmission of HCV is largely attributed to injection drug use (2). The association between injection drug use, HCV infection, and imprisonment is very close (3). People who inject drugs (PWID) account for a large proportion of the incarcerated population in the United States, Canada, Europe, and Australia (4–7), and injection drug use is prevalent during incarceration (8,9). Globally, the prevalence of HCV infection among prisoners is ≈30% (10,11). A meta-analysis of 30 studies conducted in different countries revealed a clear association between the prevalence of HCV infection among prisoners and a history of injection drug use (6).
A recent meta-analysis of HCV incidence studies among prisoners revealed a mean incidence of 16.4 (95% CI 0.8–32.1) cases per 100 person-years (11). We recently documented incidence of 14.1 (95% CI 10.0–19.3) cases per 100 person-years in 37 prisons in New South Wales (NSW), Australia, and identified recent injection drug use and Aboriginal and Torre Strait Islander descent as independent risk factors for HCV seroconversion (12). This analysis also identified high prevalence of injection drug use and sharing of injecting equipment in prisons (12). Furthermore, 13 incident cases were identified in a subcohort of 114 prisoners continuously imprisoned (i.e., without release to the community) during the study period (incidence 10.3 cases/100 person-years).
Prisons can be regarded as an enclosed network of facilities within which prisoners are frequently moved. In NSW, prisoners are often transferred between prisons (e.g., because of changes in prisoner security classifications) and temporarily moved for brief periods (e.g., to go to court or obtain medical treatment). In addition, prison sentences in Australia are typically short (average 7–9 months), but reincarceration rates are high (13).
The HCV genome evolves rapidly by mutations caused by highly error-prone replication mechanisms, which generate a swarm of constantly evolving variants (quasispecies) during every infection (14). HCV is classified into 7 genotypes and 67 subtypes (15). At the nucleotide level, each virus subtype differs by up to 25% and genotypes differ by up to 33% (16). The hypervariable region (HVR) of the HCV genome is the most variable; hence, this region is commonly used in molecular epidemiologic studies to detect clusters of persons infected via recent transmission events (17). We used sequences covering envelope (E) 1 and partial E2 (HVR1).
Acute HCV infection is largely asymptomatic; hence, the precise timing and source of transmission are usually unknown. Accordingly, virus sequencing and phylogenetic analysis have been used to reconstruct probable transmission chains from prevalent cases (18–20). Although broad linkages between HCV-infected persons have been demonstrated, previous efforts to identify probable transmission pairs among infected persons by using a combination of social network information and phylogenetic analysis techniques suggested that social and genetic distances were only weakly associated (21). By contrast, a recent report from a study that used this same approach among both prevalent and incident (newly infected) case-patients, identified probable clusters evidenced by proximity of social network and clustering analysis of core HCV sequences in a community-based cohort of PWID (22).
Our study used an integrated analysis of molecular, epidemiologic, and spatiotemporal data from a well-characterized cohort of longitudinally followed PWID. We used incident case detection in prisons to identify clusters of recent HCV transmission.
Hepatitis C Incidence and Transmission Study
The Hepatitis C Incidence and Transmission Study in Prisons (HITS-p) is a prospective study of a cohort of 498 prisoners with a history of injection drug use recruited from 37 prisons in NSW during 2005–2012 (12,23,24). At the time of preenrollment screening, all HITS-p participants were not infected with HCV; 181 subsequently became infected (12,23,24).
Study Cohort
For our study, we considered a HITS-p subset of 79 prisoners infected with HCV genotype 1 or genotype 3 for which HCV E1-HVR1 sequences were available. At ≈6-month intervals during participants’ incarceration, we collected demographic information, lifetime and follow-up risk behavior data, and blood samples for HCV serologic and virologic testing (12,23,24). These data were collected by a trained research nurse whose employment was independent of the prison system (12).
HCV Testing and Estimated Date of Infection
Blood samples were tested for presence of HCV RNA and antibodies as described elsewhere (12,23,24). For participants who had seroconverted at the incident time point (the time of sampling when a person is found to have already seroconverted), the date of infection was estimated as the midpoint between the first HCV antibody–positive and the last HCV antibody–negative test result. For participants who were HCV RNA positive but HCV antibody negative at the incident time point, the date of infection was estimated to be 51 days before the date of sampling (25).
Statistical Analyses
We used t-tests (for continuous variables) and χ2 tests (for categorical variables) to compare the demographic characteristics and risk behavior of newly infected participants with those of noninfected participants (significance level = 0.05). We used the Wilcoxon rank-sum test to assess differences in number of movements.
Sequencing of the E1-HVR1
The region encoding the last 171 bp of core, E1, and HVR1 (882 bp [nt 723–1604]) was compared with HCV strain H77 (GenBank accession no. AF009606). These sequences were then amplified by nested reverse transcription PCR as described elsewhere (26).
Phylogenetic Analysis
ClustalW (implemented in MEGA 5.2.1 [27]) was used for alignment of genotypes 1 and 3 E1-HVR1 sequences. Alignments were visually inspected and manually edited. The HKY model with gamma distribution and a proportion of invariable sites was selected as the best-fit evolutionary model by using JModelTest (28). Separate phylogenetic trees for the genotype 1 and genotype 3 alignments with a maximum-likelihood approach were generated by using PhyML (29). To check for the robustness of the trees, we performed a 1,000-bootstrap test.
Clustering Analyses
Clusters of recent HCV transmission were detected by using PhyloPart (30), a software program that identifies genetically related sequences from a given tree by use of a statistical algorithm based on analysis of pairwise patristic distances (the amount of change between any 2 sequences as depicted by the branch lengths in a phylogenetic tree). PhyloPart considers any subtree as a cluster if the median pairwise patristic distance among its members is below a set percentile threshold of the distribution of all pairwise patristic distances in the given tree (Technical Appendix).
Validation Analyses of Clusters of Recent HCV Transmission
Records for each participant (consisting of time, date, and location of entry and exit from each prison) during 2005–2012 were obtained from the NSW Department of Corrective Services. Recent HCV transmission events were validated by integrating the estimated date of infection, incarceration time and location, and the reported risk behavior of participants during follow-up in each of the phylogenetically designated clusters.
For each cluster of cases indicating recent transmission, potential transmission pairs (source and recipient) are identified as any 2 participants co-located in the same prison for at least 24 hours. The source was identified as the participant with an estimated date of infection earlier than the time of co-location with the other participant. The recipient was identified as the participant who was HCV antibody negative before co-location and who became HCV antibody positive within 12 months after co-location with the source participant. Clusters of >2 participants were considered valid with the identification of at least 1 transmission pair.
Risk behaviors (assessed prospectively during interviews at 6-month intervals) were available for the HITS-p cohort and included injection drug use and other blood-to-blood contact but excluded risks associated with sexual behavior (12). Information about drug injection and sharing of injecting equipment were obtained “since coming into prison” or “since the last interview” in association with “injected drugs,” “frequency of injecting drugs,” “use of injecting equipment after someone else,” and “frequency of use of injecting equipment.”
Participants
From 181 newly infected participants (incident case-participants) in the HIT-P cohort, 102 were excluded from the study because they were infected with an HCV genotype other than 1 or 3. The study cohort thus comprised 79 viremic incident case-participants. Most (49 [62%]) participants were male, mean ± SD age was 28 ± 7.2 years, 18 (23%) were of Aboriginal and Torre Strait Islander descent, and 61 (77%) had completed <10 years of formal education. The study cohort included 69 (87%) participants who had been previously imprisoned, and most had lifetime risk factors for blood-borne virus acquisition at baseline (Table 1). No significant differences in demographics and lifetime risk behaviors were found between the 79 study cohort participants and the 317 noninfected HITS-p cohort participants, other than previous imprisonment and having ever injected drugs while in prison (Table 1). There were no significant differences between the 79 study cohort participants and the 102 excluded infected participants (Table 1).
Phylogenetics
Figure 1
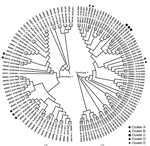
Figure 1. Phylogenetic trees composed of 129 sequences from 79 participants infected with hepatitis C virus genotypes (gt) 1a, 1b, or 3a, New South Wales, Australia, 2005–2012. Names on the tips of the...
A total of 129 sequences of E1-HVR1 were obtained from the 79 participants; 26 participants were infected with HCV genotype 1a, 5 with genotype 1b, 44 with HCV genotype 3a, and 4 with HCV genotypes 1a and 3a at different times. These reinfection cases were included in both the genotype 1 and genotype 3 analyses with the corresponding genotype-specific sequences. For participants infected with genotype 1, sequences were available from 1 viremic time point for 19 participants, from 2 time points for 10, and from 3 time points for 6. For participants infected with genotype 3, sequences were available from 1 viremic time point for 28 participants, from 2 time points for 15, and from 3 time points for 5. Phylogenetic trees were constructed for the genotype 1 and genotype 3 E1-HVR1 sequences (Figure 1).
Clustering
Figure 2

Figure 2. Analysis of hepatitis C virus transmission clusters identified across a range of percentile thresholds among prisoners in New South Wales, Australia, 2005–2012. Analysis shows the relationship between the number of clusters...
The optimal cutoff patristic distance designating recent transmission clusters was determined first by investigation of a range of percentile thresholds from the distribution of pairwise patristic distances (Technical Appendix Methods). As expected at the minimum percentile value, only within-participant clusters were detected, while at the maximum, all sequences for each genotype were included in a single between-participant cluster (Figure 2). On this basis, the chosen cutoff patristic distance for designation of between-participant clusters was 0.099 for genotype 1 and 0.095 for genotype 3 (corresponding to 0.034 and 0.022 nt substitutions/site in the E1-HVR1 region, respectively).
Figure 3

Figure 3. Analysis of pairwise patristic distances between hepatitis C virus sequences from the same participant (within-participant) sampled over time, and from between participants also sampled over time, among prisoners in New South...
To assess the effect of the time interval between sampling points on the distribution of pairwise patristic distances, and hence the designated thresholds, we studied the relationship between the time of collection and the pairwise patristic distance between all the sequences available for the study cohort (longitudinally within-participant and between-participant). The pairwise patristic distances between hosts was independent of the time interval (Figure 3). The degree of viral divergence reflected by patristic distances among sequences from within the same participant increased with the time interval between the collection time points. Within the time window analyzed (up to 4 years), within-participant genetic distances remained smaller than those from between-participant pairs. Only a small proportion of the between-participant genetic distances were within the range of within-participant pairs.
Further validation analyses including sequences from a single-source HCV outbreak (Technical Appendix Results 1) showed that within-participant evolution could generate patristic distances greater than those observed between the sequence of the source and infected recipients when collected up to 23 years after transmission. However, the median distribution of these distances revealed that between-participant distances were significantly higher than within-participant differences.
Last, to assess the potential effect of virus diversity within the quasispecies of a single-source host and the potential transmission of a minor variant to a new recipient, the distribution of pairwise patristic distances between all E1-HVR1 variants within the quasispecies from 2 time points collected over 1 year from 2 participants followed from primary HCV infection was analyzed to a sensitivity of variants representing 1% of the quasispecies (Technical Appendix Results 2). Again, the maximum within-participant genetic distance within the quasispecies did not exceed the genetic distances between consensus sequences identified in between-participant analyses.
Clusters of Recent Transmission and Spatiotemporal Validation
One cluster of recent transmission was detected among 57 genotype 1 sequences (Figure 1, cluster A). This cluster consisted of 3 participants (nos. 117, 461, and 315); median pairwise patristic distance was 0.058. Two clusters were detected among genotype 3 sequences. The first (Figure 1, cluster B) consisted of 2 participants (nos. 304 and 357); median pairwise patristic distance was 0.011. The second cluster (Figure 1, cluster C) consisted of 2 participants (nos. 426 and 302); median pairwise patristic distance was 0.090. Two more clusters were detected just above the designated patristic distance cutoff (Technical Appendix Results 3). The estimated date of infection, incarceration time and location, and reported risk behavior for each cluster member were analyzed to provide convergent evidence for likely transmission events (Table 2).
Figure 4
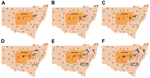
Figure 4. Reconstruction of the likely hepatitis C virus transmission dynamics among prisoners in New South Wales, Australia, 2005–2012. Geographic representation of the transmission dynamics among 3 participants identified in cluster A over...
Video 1
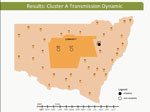
Video 1. Hepatitis C virus transmission dynamics among prisoners in cluster A, involving participants 315, 117, and 461, New South Wales, Australia, 2005–2012.
Video 2
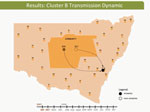
Video 2. Hepatitis C virus transmission dynamics among prisoners in cluster B, involving participants 304 and 357, New South Wales, Australia, 2005–2012.
Video 3
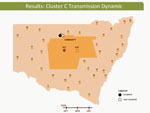
Video 3. Hepatitis C virus transmission dynamics among prisoners in cluster C, involving participants 302 and 426, New South Wales, Australia, 2005–2012.
These dynamic participant movements were reconstructed for each transmission cluster. In cluster A, HCV was likely to have been transmitted from participant 315 to participants 117 and 461 (Figure 4). The estimated date of infection with genotype 1a for participant 315 was October 30, 2007; this participant had been in the same prison as participant 117 for 22 days (December 31, 2007–January 22, 2008). Both participants reported injecting drugs and sharing injecting equipment during the period of co-location. Participant 117 was then found to be viremic with genotype 1a in a sample obtained on August 20, 2008, giving an estimated date of infection of February 27, 2008. In another likely transmission event, participant 315 had been in the same prison with participant 461 on 2 occasions: for 13 days (June 29–July 11, 2008) and for 9 days (September 24–October 1, 2008). Both participants reported injecting drugs and sharing injecting equipment during the period of co-location. Participant 461 was then found to be viremic with genotype 1a according to a sample dated November 3, 2008; estimated date of infection was October 6, 2008 (Video 1, http://wwwnc.cdc.gov/EID/article/21/5/14-1832-F1.htm). In transmission cluster B, HCV was likely to have been transmitted from participant 304 to 357. Estimated date of infection with genotype 3 for participant 304 was March 17, 2007; this participant had been in the same prison with participant 357 for 28 days, October 26–November 23, 2007. Both participants reported injecting drugs (although participant 304 did not report sharing injecting equipment) during the period of co-location. Participant 357 was then found to be viremic with genotype 3 according to a sample dated April 17, 2009; estimated date of infection was September 11, 2008 (Video 2, http://wwwnc.cdc.gov/EID/article/21/5/14-1832-F2.htm). In transmission cluster C, HCV genotype 3 was likely to have been transmitted from participant 302 to participant 426. Estimated date of infection for participant 302 was October 11, 2007; this participant had been in the same prison with participant 426 for 9 days, December 9–18, 2008. Both participants reported injecting drugs and sharing injecting equipment during this period of co-location. Participant 426 was then found to be viremic according to a sample obtained on July 9, 2009; estimated date of infection was December 21, 2008 (Video 3, http://wwwnc.cdc.gov/EID/article/21/5/14-1832-F3.htm). Of note, participant 302 is female, and participant 426 is male. Despite the short period of co-location, it is unlikely that prisoners of different sex could interact directly in the prisons, although shared use of a single injection device may have been possible.
Relationship between Phylogenetic Clustering and Movement Dynamics
In NSW, a high number of prisoner movements are common; prisoners are often transferred between correctional centers or released to the outside community. During the study period (2005–2012), participants from the HITS-p cohort were moved to a different location (a prison or the outside community) a mean of 17 times (Technical Appendix Table 2), and the 79 participants in the study cohort moved a mean (± SD) of 22 ± 13.55 times, with a mean of 4 ± 2.83 release events. The 7 participants from the 3 clusters of recent HCV transmission moved to a different location a mean of 28 ± 15.75 times, a significantly greater number of times than for the HITS-p cohort as a whole (p = 0.002) and for the subcohort of uninfected participants (p<0.001). These differences remained significant when movements from one prison to another and release to outside community were tested separately (p<0.05 for all).
Our molecular epidemiology analysis combined with detailed spatiotemporal and behavioral risk data identified several clusters of recent transmission of HCV infection within NSW prisons. This study shows direct evidence of ongoing HCV transmission among PWID in a prison setting.
Previous phylogenetic studies have examined associations between HCV infection and risk and demographic characteristics, including injection drug use (17,21,22,31,32). Moreover, those studies have defined transmission clusters with a threshold value fixed a priori, such as a maximum genetic distance of 2%–5% (17), or with a bootstrap cutoff value (22). Here, an empirically optimized threshold, which can also be larger than the typical threshold fixed in previous studies, was used to search for clusters of recent transmission exclusively among incident case-participants.
Despite a high prevalence of chronic HCV infection in prison populations, 3 clusters of transmission were identified in phylogenetic analysis of only 79 participants with recent HCV infection identified during 2005–2012. During this period, ≈20,000 persons were imprisoned annually in NSW; HCV antibody prevalence was ≈30% (33,34), which equates to ≈4,500 persons with chronic HCV infection (assuming 25% of those cleared infection) who were imprisoned annually. When discounted for 40% recidivism (13), this calculation yields ≈19,000 infected prisoners who may have acted as sources for HCV transmission over the study period. In our analysis, the numbers of movements were higher among newly infected participants than among noninfected participants, suggesting that transmission is associated with frequent movements between prisons and from prison to the outside community. Such frequent movements could increase the chance of contact with infected persons or could be otherwise associated with behavior that puts a person at increased risk for HCV transmission.
It is possible that recently infected participants are more likely than chronically infected participants to transmit infection (35). This possibility could result from higher infectivity of the transmitted founder viruses, which are intrinsically adapted for successful transmission and dominate the acute phase of infection (14). In contrast, a high circulating viral load is associated with an increased probability of vertical HCV transmission (36,37). However, in our study of PWID, the viral loads (recorded in the blood samples close to the time of transmission) in the source case-participants in the clusters were only low to moderate (data not shown). An alternative explanation is the possibility that these clusters are part of an existing network of high-risk PWID across prisons.
The genetic diversity between variants within the quasispecies during a single infection can become substantial because of the high mutation rate of the virus and the selection pressures of the host immune response. This diversity could influence transmission events because a minor variant in the source can be preferentially transmitted and then dominate the virus population in the recipient host. Therefore, consensus sequencing might not be sufficient for detection of clusters in which transmission is driven by rare variants. Despite the fact that the maximum genetic distances observed within the quasispecies in the selected samples studied here did not exceed the mean genetic distance between hosts, it remains possible that additional transmission clusters may have become evident had this approach been used for all samples.
Our study has several limitations. First, the virus populations involved in transmission events occurring several months after infection might differ from those involved in the acute phase of infection because of the rapid diversification of the virus genome. Therefore, these findings may underestimate ongoing transmission in prisons. Second, although the viruses infecting persons in the clusters were closely related, there is a possibility that unknown participants outside the cohort were also part of the transmission chains; hence, the identified recipient could have been infected by an intermediary source. This possibility may be relevant to probable indirect transmission of HCV from a female participant to a male participant in cluster C because male and female prisoners are segregated in prisons in Australia. Third, because the proposed method uses information collected only during incarceration, data on injecting and sharing behavior in the outside community were not available. Indeed, only 20 (25%) prisoners in the study cohort were continuously imprisoned in the 6 months before the estimated date of infection. Finally, risk behavior could have been underestimated because of the underreporting of sensitive and socially stigmatized behavior during interviews.
From a global perspective, public health control programs have had relatively limited effects on mitigating HCV transmission. The analysis of the HITS-p cohort showed that opioid substitution therapy uptake reaches only 20% of the population (12,24), despite 64% reporting having ever injected heroin. A recent study on a cohort of PWID in NSW has identified a strong protective effect of opioid substitution therapy (38). The combination of needle and syringe exchange programs and opioid substitution therapy programs is the most effective approach for mitigating HCV transmission, reducing incidence by a substantial amount (30%–80%) (39,40). However, needle and syringe exchange programs remain prohibited in NSW prisons. By identifying ongoing HCV transmission in prisons, this study advocates for new strategies for reducing risk behavior, such as increasing opioid substitution therapy use and eventually introducing needle and syringe programs in prison settings.
Mr. Bretaña is a PhD candidate in the Inflammation and Infection Research Centre, School of Medical Sciences, The University of New South Wales, Sydney, Australia. His research interests are phylogenetics, epidemiology, and computational modeling of hepatitis C virus transmission.
Acknowledgments
The HITS-p investigators include Kate Dolan, Paul Haber, William Rawlinson, Carla Treloar, Greg Dore, Lisa Maher, and authors Andrew Lloyd and Fabio Luciani.
This work was supported by grants from National Health and Medical Research Council of Australia including NSW Health, Justice Health, and Corrective Services NSW as partners (grant nos. 222887 and 1016351); and by a National Health and Medical Research Council of Australia Practitioner Fellowship (grant no. 1043067 to A.L.)
References
- Kretzer IF, do Livramento A, da Cunha J, Goncalves S, Tosin I, Spada C, Hepatitis C worldwide and in Brazil: silent epidemic—data on disease including incidence, transmission, prevention, and treatment. The Scientific World Journal. 2014;2014: article ID 827849. DOIGoogle Scholar
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–67. DOIPubMedGoogle Scholar
- Hunt DR, Saab S. Viral hepatitis in incarcerated adults: a medical and public health concern. Am J Gastroenterol. 2009;104:1024–31 . DOIPubMedGoogle Scholar
- Harrison PM, Beck AJ. Prisoners in 2005. Bureau of Justice Statistics Bulletin [cited 2013 Oct 24]. http://www.bjs.gov/content/pub/pdf/p05.pdf
- Government of Canada. Corrections and conditional release statistical overview [cited 2013 Oct 24]. http://publications.gc.ca/collections/Collection/PS4-12-2004E.pdf
- Vescio MF, Longo B, Babudieri S, Starnini G, Carbonara S, Rezza G, Correlates of hepatitis C virus seropositivity in prison inmates: a meta-analysis. J Epidemiol Community Health. 2008;62:305–13. DOIPubMedGoogle Scholar
- Butler TM, The L. 2001 New South Wales inmate health survey [cited 2013 Nov 1]. http://www.nobars.org.au/downloads/Inmate_Health_Survey_2001.pdf
- Kinner SA, Jenkinson R, Gouillou M, Milloy MJ. High-risk drug-use practices among a large sample of Australian prisoners. Drug Alcohol Depend. 2012;126:156–60. DOIPubMedGoogle Scholar
- Pollini RA, Alvelais J, Gallardo M, Vera A, Lozada R, Magis-Rodriquez C, The harm inside: injection during incarceration among male injection drug users in Tijuana, Mexico. Drug Alcohol Depend. 2009;103:52–8. DOIPubMedGoogle Scholar
- Larney S, Kopinski H, Beckwith CG, Zaller ND, Jarlais DD, Hagan H, Incidence and prevalence of hepatitis C in prisons and other closed settings: results of a systematic review and meta-analysis. Hepatology. 2013;58:1215–24. DOIPubMedGoogle Scholar
- Luciani F, Bretana NA, Teutsch S, Amin J, Topp L, Dore GJ, A prospective study of hepatitis C incidence in Australian prisoners. Addiction. 2014;109:1695–706. DOIPubMedGoogle Scholar
- Payne J. Recidivism in Australia: findings and future research [cited 4 Nov 2013]. http://www.aic.gov.au/documents/0/6/B/%7B06BA8B79-E747-413E-A263-72FA37E42F6F%7Drpp80.pdf
- Bull RA, Luciani F, McElroy K, Gaudieri S, Pham ST, Chopra A, Sequential bottlenecks drive viral evolution in early acute hepatitis C virus infection. PLoS Pathog. 2011;7:e1002243. DOIPubMedGoogle Scholar
- Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology. 2014;59:318–27. DOIPubMedGoogle Scholar
- Simmonds P, Bukh J, Combet C, Deleage G, Enomoto N, Feinstone S, Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–73 . DOIPubMedGoogle Scholar
- Jacka B, Lamoury F, Simmonds P, Dore GJ, Grebely J, Applegate T. Sequencing of the hepatitis C virus: a systematic review. PLoS ONE. 2013;8:e67073. DOIPubMedGoogle Scholar
- Féray C, Bouscaillou J, Falissard B, Mohamed MK, Arafa N, Bakr I, A novel method to identify routes of hepatitis C virus transmission. PLoS ONE. 2014;9:e86098. DOIPubMedGoogle Scholar
- Matthews GV, Pham ST, Hellard M, Grebely J, Zhang L, Oon A, Patterns and characteristics of hepatitis C transmission clusters among HIV-positive and HIV-negative individuals in the Australian trial in acute hepatitis C. Clin Infect Dis. 2011;52:803–11. DOIPubMedGoogle Scholar
- Phylogenetic clustering of hepatitis C virus among people who inject drugs in Vancouver, Canada. Hepatology. 2014;60:1571–80 Jacka B and Applegate T and Krajden M and Olmstead A and Harrigan PR and Marshall BD. DOIPubMedGoogle Scholar
- Aitken CK, McCaw RF, Bowden DS, Tracy SL, Kelsall JG, Higgs PG, Molecular epidemiology of hepatitis C virus in a social network of injection drug users. J Infect Dis. 2004;190:1586–95. DOIPubMedGoogle Scholar
- Sacks-Davis R, Daraganova G, Aitken C, Higgs P, Tracy L, Bowden S, Hepatitis C virus phylogenetic clustering is associated with the social-injecting network in a cohort of people who inject drugs. PLoS ONE. 2012;7:e47335. DOIPubMedGoogle Scholar
- Dolan K, Teutsch S, Scheuer N, Levy M, Rawlinson W, Kaldor J, Incidence and risk for acute hepatitis C infection during imprisonment in Australia. Eur J Epidemiol. 2010;25:143–8. DOIPubMedGoogle Scholar
- Teutsch S, Luciani F, Scheuer N, McCredie L, Hosseiny P, Rawlinson W, Incidence of primary hepatitis C infection and risk factors for transmission in an Australian prisoner cohort. BMC Public Health. 2010;10:633. DOIPubMedGoogle Scholar
- Page-Shafer K, Pappalardo BL, Tobler LH, Phelps BH, Edlin BR, Moss AR, Testing strategy to identify cases of acute hepatitis C virus (HCV) infection and to project HCV incidence rates. J Clin Microbiol. 2008;46:499–506. DOIPubMedGoogle Scholar
- Pham ST, Bull RA, Bennett JM, Rawlinson WD, Dore GJ, Lloyd AR, Frequent multiple hepatitis C virus infections among injection drug users in a prison setting. Hepatology. 2010;52:1564–72. DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. DOIPubMedGoogle Scholar
- Darriba DT, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012.9:772.
- Guindon SG. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704 Gascuel O. DOIPubMedGoogle Scholar
- Prosperi MC, Ciccozzi M, Fanti I, Saladini F, Pecorari M, Borghi V, A novel methodology for large-scale phylogeny partition. Nat Commun. 2011;2:321.
- Pilon R, Leonard L, Kim J, Vallee D, De Rubeis E, Jolly AM, Transmission patterns of HIV and hepatitis C virus among networks of people who inject drugs. PLoS ONE. 2011;6:e22245. DOIPubMedGoogle Scholar
- Hope VD, Hickman M, Ngui SL, Jones S, Telfer M, Bizzarri M, Measuring the incidence, prevalence and genetic relatedness of hepatitis C infections among a community recruited sample of injecting drug users, using dried blood spots. J Viral Hepat. 2011;18:262–70. DOIPubMedGoogle Scholar
- The University of New South Wales. HIV/AIDS, viral hepatitis and sexually transmissible infections. In: 2009 annual surveillance report [cited 2014 Sep 2]. http://kirby.unsw.edu.au/sites/default/files/hiv/resources/2009AnnualSurvReport.pdf
- Butler T, Papanastasiou C. National prison entrants’ bloodborne virus and risk behaviour survey 2004 and 2007 [cited 2014 Oct 20]. http://ndri.curtin.edu.au/local/docs/pdf/publications/R223.pdf
- Thomas DL. Global control of hepatitis C: where challenge meets opportunity. Nat Med. 2013;19:850–8. DOIPubMedGoogle Scholar
- Paternoster DM, Santarossa C, Grella P, Palu G, Baldo V, Boccagni P, Viral load in HCV RNA–positive pregnant women. Am J Gastroenterol. 2001;96:2751–4. DOIPubMedGoogle Scholar
- Ruiz-Extremera A, Munoz-Gamez JA, Salmeron-Ruiz MA, de Rueda PM, Quiles-Perez R, Gila-Medina A, Genetic variation in interleukin 28B with respect to vertical transmission of hepatitis C virus and spontaneous clearance in HCV-infected children. Hepatology. 2011;53:1830–8. DOIPubMedGoogle Scholar
- White B, Dore GJ, Lloyd AR, Rawlinson WD, Maher L. Opioid substitution therapy protects against hepatitis C virus acquisition in people who inject drugs: the HITS-c study. Med J Aust. 2014;201:326–9. DOIPubMedGoogle Scholar
- Palmateer N, Kimber J, Hickman M, Hutchinson S, Rhodes T, Goldberg D. Evidence for the effectiveness of sterile injecting equipment provision in preventing hepatitis C and human immunodeficiency virus transmission among injecting drug users: a review of reviews. Addiction. 2010;105:844–59. DOIPubMedGoogle Scholar
- Turner KM, Hutchinson S, Vickerman P, Hope V, Craine N, Palmateer N, The impact of needle and syringe provision and opiate substitution therapy on the incidence of hepatitis C virus in injecting drug users: pooling of UK evidence. Addiction. 2011;106:1978–88. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1Additional HITS-p investigators are listed at the end of this article.
Table of Contents – Volume 21, Number 5—May 2015
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Fabio Luciani, Inflammation and Infection Research Center, School of Medical Sciences, The University of New South Wales, Sydney, New South Wales, Australia; email
Top