Volume 21, Number 5—May 2015
CME ACTIVITY - Perspective
Recent US Case of Variant Creutzfeldt-Jakob Disease—Global Implications
Introduction
![]()
Medscape, LLC is pleased to provide online continuing medical education (CME) for this journal article, allowing clinicians the opportunity to earn CME credit.
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint providership of Medscape, LLC and Emerging Infectious Diseases. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)TM. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/eid; (4) view/print certificate.
Release date: April 15, 2015; Expiration date: April 15, 2016
Learning Objectives
Upon completion of this activity, participants will be able to:
• Describe the clinical presentation of variant Creutzfeldt-Jakob disease, based on a case report and review
• Discuss diagnostic testing for variant Creutzfeldt-Jakob disease
• Determine the global implications of this report of a new US case of variant Creutzfeldt-Jakob disease
CME Editor
Karen L. Foster, Technical Writer/Editor, Emerging Infectious Diseases. Disclosure: Karen L. Foster has disclosed no relevant financial relationships.
CME Author
Laurie Barclay, MD, freelance writer and reviewer, Medscape, LLC. Disclosure: Laurie Barclay, MD, has disclosed no relevant financial relationships.
Authors
Disclosures: Atul Maheshwari, MD, Michael P. Fischer, MD, MPH, TM; Alicia Parker, MD; Aarthi Ram, MD; Luis Concha-Marambio, BS; Yvonne Cohen, BS; Ermias D. Belay, MD; Ryan A. Maddox, PhD, MPH; Simon Mead, MD, PhD; J. Clay Goodman, MD; Joseph S. Kass, MD, JD; Lawrence B. Schonberger, MD, MPH; and Haitham M. Hussein, MD, MSc, have disclosed no relevant financial relationships. Pierluigi Gambetti, MD, has disclosed the following relevant financial relationships: served as an advisor or consultant to and received grants for clinical research from Ferring Pharmaceuticals. Claudio Soto, PhD, has disclosed the following relevant financial relationships: owns stock, stock options, or bonds from and is founder and Chief Scientific Officer of Amprion Inc.
This article may discuss off-label uses of drugs, mechanical devices, biologics, or diagnostics approved by the Food and Drug Administration (FDA) for use in the United States and investigational drugs, mechanical devices, biologics, or diagnostics not approved by the FDA for use in the United States.
Abstract
Variant Creutzfeldt-Jakob disease (vCJD) is a rare, fatal prion disease resulting from transmission to humans of the infectious agent of bovine spongiform encephalopathy. We describe the clinical presentation of a recent case of vCJD in the United States and provide an update on diagnostic testing. The location of this patient’s exposure is less clear than those in the 3 previously reported US cases, but strong evidence indicates that exposure to contaminated beef occurred outside the United States more than a decade before illness onset. This case exemplifies the persistent risk for vCJD acquired in unsuspected geographic locations and highlights the need for continued global surveillance and awareness to prevent further dissemination of vCJD.
Prion disorders are a unique class of diseases caused by pathologically misfolding proteins leading to neurodegeneration (1). Creutzfeldt-Jakob disease (CJD), one such prion disorder, is divided into 4 etiologic categories: sporadic (sCJD), familial, iatrogenic, and variant (vCJD). CJD is incurable and inevitably fatal.
The emergence of vCJD was linked to an earlier epidemic of bovine spongiform encephalopathy (BSE) in the United Kingdom through the consumption of contaminated beef (2). Since the first report in 1996, a total of 229 vCJD cases have been reported worldwide: 177 in the United Kingdom; 27 in France; and 25 distributed in 10 other countries, including the United States (3). Incidence of vCJD peaked in the United Kingdom and France in 1999 and 2004, respectively. Countries outside of the United Kingdom were apparently affected through importation of beef and live cattle from the United Kingdom (3,4). Only 5 vCJD cases have previously been reported in North America: 3 in the United States and 2 in Canada. All 5 patients had lived in either the United Kingdom (3 patients) or the Kingdom of Saudi Arabia (2 patients), prompting the conclusion that vCJD was acquired overseas rather than in the United States or Canada (5).
We report the fourth confirmed case of vCJD in the United States and note that, unlike for the previous 3 cases, the specific country in which this BSE/vCJD infection was acquired is less clear. We found no definite epidemiologic link to a country where other known vCJD patients probably had been infected. We also review the clinical features and diagnostic challenge of vCJD, with special consideration of the possible public health concerns and global risks inherent to this disease.
The patient was a man in his forties who was born and raised in the Middle East. He lived in Russia for several years while completing a professional degree and briefly returned to the Middle East before taking up his final residence in the United States during the late 1990s.
Family members, friends, and co-workers were interviewed. The Texas Department of State Health Service’s investigation ascertained and verified that he had resided in Lebanon, Kuwait, Russia, and the United States. The patient’s family affirmed that, if the man had traveled to European countries, that travel was brief and infrequent. We found no evidence indicating that he had ever stayed in Great Britain, France, Ireland, or Saudi Arabia.
Figure 1

Figure 1. Timeline of course of illness and major diagnostic tests for US patient with variant Creutzfeldt-Jakob disease. CT, computed tomography; EEG, electroencephalography; MRI, magnetic resonance imaging; LP, lumbar puncture..
The patient’s family and co-workers described him as healthy and high-functioning before illness onset. The condition first manifested in late 2012 as depression and anxiety. These symptoms were initially subtle and did not interfere with his daily activities. Delusions and hallucinations were observed shortly after the onset of illness and in subsequent months were associated with changes in behavior in the form of withdrawal, isolation, secrecy, aggression, poor judgment, and lack of insight (Figure 1).
Intermittent numbness and paresthesias of the left face and upper extremity began in the fifth month of illness. These symptoms were attributed to a motor vehicle collision within the previous weeks. A few months later, when the symptoms started to affect the contralateral side, brain and cervical spine magnetic resonance imaging (MRI) was performed and showed multilevel degenerative changes and spinal foraminal stenosis, leading to the conclusion that most of his signs and symptoms were caused by cervical disc disease. The etiology for his facial paresthesias remained unclear.
The patient’s psychiatric condition gradually continued to worsen, and he was hospitalized multiple times in the 13th and 14th months after illness onset. Several diagnoses were entertained, including depression with psychotic features and bipolar disorder with psychosis. Despite treatment of symptoms, his condition continued to deteriorate.
Figure 2

Figure 2. Magnetic resonance imaging (MRI) results for a US patient with variant Creutzfeldt-Jakob disease. T1 sequence (A) and T2 FLAIR sequence (B) show the “pulvinar” or “hockey stick” sign (arrowhead). Initial diffusion...
Because of increasing agitation, the patient was once again brought to the emergency department in the 14th month after symptom onset, where the neurology service was consulted. He was restless, irritable, disinhibited, and impulsive. He exhibited choreiform movements, most pronounced in the left upper extremity, in addition to myoclonus and ataxia. Reflexes were reduced throughout the upper and lower extremities. MRI of the brain showed mild diffuse volume loss, T2 hyperintensity with subtle restricted diffusion in the pulvinar nuclei of bilateral thalami (pulvinar sign), and subtle restricted diffusion in the right frontal cortex (cortical ribbon sign) (arrows) (Figure 2). Analysis of cerebrospinal fluid (CSF) showed elevated protein at 120 mg/dL (reference range 15–60 mg/dL) and a normal cell count, prompting a course of intravenous steroids and plasmapheresis for suspected autoimmune encephalitis.
Extensive investigations for infectious and autoimmune encephalitides, malignancy, heavy metal intoxication, vitamin deficiency, and rheumatologic and endocrine disorders were concomitantly pursued. CSF was sent to the National Prion Disease Pathology Surveillance Center (NPDPSC; Cleveland, OH, USA) for evaluation of 14-3-3 and Tau protein levels, as well as real-time quaking-induced conversion (RT-QuIC) testing for prion detection (Table 1).
During the first several weeks after admission, the patient’s behavior and chorea improved, which was interpreted as a possible response to immunomodulatory therapy. He therefore underwent further intravenous immunoglobulin treatment and received 2 doses of rituximab. After a few weeks of a relatively stationary course, his condition deteriorated. Dysarthria, truncal ataxia, and lower extremity weakness developed, with subsequent loss of his ability to ambulate.
After the laboratory tests for antibody-mediated autoimmune and paraneoplastic encephalidites yielded negative results, “probable vCJD” was diagnosed. The Texas Department of State Health Service and the Centers for Disease Control and Prevention (Atlanta, GA, USA) were notified of the suspected case. Samples of blood, urine, and CSF were sent to the NPDPSC and the Medical Research Council Prion Unit (London, UK). Brain and tonsil biopsies were deferred because of concern that the patient would not tolerate either biopsy procedure.
By the 16th month of illness, left arm plegia and a severe bulbar palsy developed. Within 1 month, the patient became akinetic-mute and entirely bedridden. After the third episode of aspiration pneumonia, sepsis developed. After discussion with the family, goals of care were restricted to comfort measures only. The patient died shortly thereafter, almost 18 months after initial onset of symptoms.
Figure 3
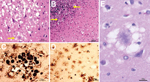
Figure 3. Results of histopathologic and immunohistochemical analyses for a US patient with variant Creutzfeldt-Jakob disease. A) Hematoxylin and eosin staining shows many typical florid plaques (A, arrow) occasionally forming clusters; large vacuole...
A brain-only autopsy was performed. Conventional histologic examination demonstrated numerous typical florid plaques that occasionally formed clusters and were often mixed with patches of spongiform change made of large vacuoles (Figure 3, panel A). This lesion pattern was present throughout the cerebral cortex, except for the hippocampal formation and lower temporal gyri. Basal ganglia and thalamus showed severe spongiform change with only scattered plaques. Prion plaques, but not well-formed florid plaques, were present in the granule cell layer of the cerebellum (Figure 3, panel B).
Prion protein immunostaining showed intense immunoreaction of the core of the florid plaques and patchy and granular deposits that were arranged in rounded clusters (Figure 3, panel C). In addition, the immunostaining highlighted cells with short processes stemming from the round perikaryon in a spoke wheel–like fashion (Figure 3, panel D). These cells were observed especially in the cerebral cortex and molecular layer of the cerebellum, where plaques and plaque-like deposits were also present. Clusters of intensely staining kuru plaques were seen in the granule cell layer of the cerebellum.
Figure 4

Figure 4. Results of biochemical testing of a US patient with variant Creutzfeldt-Jakob disease (vCJD). A) Immunoblot of vCJD patient and controls. All the PK-treated preparations show similar electrophoretic profiles characterized by 3...
Western blot showed the typical electrophoretic pattern of the protease-treated scrapie prion protein (PrPSc) associated with vCJD. This pattern, commonly identified as 2B, is characterized by the 19 kDa molecular weight of the unglycosylated band of PrPSc after proteinase K digestion, unlike type 1, in which the size of the unglycosylated band is 21 kDa (Figure 4, panel A). In addition, the distribution of the PrPSc glycoforms differs; the diglycosylated form is the predominant form in vCJD, whereas the most represented band in sCJD is the monoglycosylated form.
Urine samples from this patient were collected before death and subjected to protein misfolding cyclic amplification (PMCA) analysis. Samples were tested twice, and both times the assay results were positive (Figure 4, panel B). Therefore, on the basis of histopathologic and biochemical analysis, the diagnosis of vCJD in the present case is definitive.
When a rare disease manifests with a common symptom, that makes the diagnosis even more difficult. Unlike sCJD, which often presents with the relatively rare symptom of rapidly progressing dementia, vCJD usually presents with psychiatric symptoms, which are relatively common. Because of these difficulties, diagnosis is usually delayed. Development of other neurologic manifestations (e.g., chorea, ataxia) supports a diagnosis of sCJD or vCJD. Painful sensory complaints can be a particularly helpful clue pointing to vCJD because they are generally not reported with sCJD. In addition, vCJD onset occurs at a younger age than does sCJD (mean 26 vs. 65 years), and disease course of vCJD is longer than that of sCJD (mean 14 months vs. 4 months) (7,8). Despite the statistical differences in onset and duration, the distinction between sCJD and vCJD is not always clear (9). In 1 study, 13 sCJD patients with psychiatric presentation had clinical characteristics more similar to vCJD than sCJD (younger age at onset and longer survival), and depression was the most common presenting symptom (10). Regardless, in the patient reported here, clinical onset and duration of illness were typical of vCJD. The clinical differentiation between sCJD and vCJD is reflected by the World Health Organization diagnostic criteria (7,8,11,12) (Table 2).
Three well-known diagnostic tests can point the clinician to sCJD: MRI (diffusion restriction and T2 hyperintensity of the cortex and basal ganglia), electroencephalography (periodic sharp wave complexes), and CSF analysis (elevated 14-3-3/Tau levels) (13). In contrast, for vCJD, electroencephalography and CSF analysis do not show any specific changes. The only positive finding of diagnostic value in vCJD from these tests is the pulvinar sign on MRI, which is not entirely specific for vCJD and can be absent in up to 9% of cases, even after multiple MRIs (14). According to World Health Organization criteria, the only way to diagnose “definite vCJD” while the patient is living is a brain biopsy (15). In contrast to sCJD, lymphoid tissue in vCJD has a high detection rate of PrPSc (16), so tonsil biopsy has become an alternative to brain biopsy, with recent investigations showing high sensitivity and specificity (17).
The panel of diagnostic tests for CJD used for many years at the NPDPSC includes level determinations of 14-3-3 and Tau proteins in the CSF. These tests are highly sensitive for sCJD, the most common form of human prion disease, but not for vCJD. A negative 14-3-3 has a negative predictive value of 63%, and a negative Tau has a negative predictive value of 81% (18). If both tests are negative, the negative predictive value for vCJD rises to 84% (18), which leaves ≈3 of 20 patients with a false-negative result, as in the patient reported here.
RT-QuIC, a relatively new test available at the NPDPSC, can detect minute amounts of prions in CSF by amplifying PrPSc using recombinant PrP as substrate. If positive, RT-QuIC is very specific for diagnosing sCJD, but it has been reported to be negative in vCJD (3), as in the CSF sample of the patient reported here.
Analysis of the PrP gene coding regions demonstrating the presence of methionine homozygosity at codon 129 has been found in all cases of proven primary vCJD examined (3,19), including in the case reported here. However, because >40% of the population can be methionine homozygous, sequencing of PrP gene coding regions is helpful only in providing supportive evidence for increased susceptibility to prion disease (20).
One experimental test for vCJD is a blood-based direct detection assay that takes advantage of the high affinity of PrPSc for metal surfaces to capture and immunodetect the minute amounts of PrPSc expected to be present in the blood of vCJD patients (21). Application of this test on specimens collected from >5,000 persons did not indicate false-positive findings; however, the sensitivity reached only 71% (22) and was negative in the patient in our report.
Another new experimental test is based on PrPSc detection in urine. This test uses PMCA technology to amplify and then visualize traces of PrPSc that might be present in the patient’s urine and has nearly 93% sensitivity and 100% specificity (6). Although the high accuracy of the assay might decrease when larger cohorts are examined, this entirely noninvasive test is likely to be useful in screening for vCJD. This test was the only one conducted on a sample obtained before the death of the patient reported here that yielded a positive result. PrPSc was detected in this patient only after 1 round of 96 PMCA cycles, indicating that the amount of PrPSc in urine was higher than that in most of the other vCJD patients analyzed (6).
The neuropathology of vCJD is distinct from that of sCJD, and the classification system for neuropathologic subtypes of sCJD has been described in detail elsewhere (23–25). The diagnosis of vCJD was definitively confirmed in this case by the postmortem histologic and PrPSc examinations, which demonstrated the widespread presence of florid plaques, the typical electrophoretic profile of the PrPSc (3), and the PrP immunostaining demonstrating rounded cells surrounded by short delicate processes resulting in a feathery appearance (26). The identity of these cells remains to be determined.
This case highlights several diagnostic challenges presented by vCJD. Studies on a vCJD cohort from the United Kingdom have shown a mean interval of 2.5 months between clinical onset and the first medical examination. The overall average delay between onset and suspicion of vCJD is 8.9 months (7). This case met similar delays, despite the typical clinical presentation (3).
The significantly elevated protein concentration in the CSF in this patient, up to 204 mg/dL, was a confounder. CSF protein in vCJD is usually normal; the highest concentration previously reported was 90 mg/dL (19). We remain unsure about the source of this finding. The cortical ribbon sign on MRI, although a common finding in sCJD, was also not previously reported in vCJD (14).
Another major difficulty in this case was proving the diagnosis with a high level of certainty before death so the goals of care could be adjusted accordingly. Despite our clinical suspicion, results of all the initial laboratory tests were negative. The pulvinar sign, which is relatively sensitive and specific for vCJD (14), did not exclude other disorders that rarely have similar features, including Wernicke’s encephalopathy and inflammatory limbic encephalitis (27–29). By the time brain or tonsil biopsy was considered, the patient was considered unable to tolerate the procedure. The only positive test specific for vCJD in this patient was PrPSc detection in urine using the PMCA test. Unfortunately, the result became available only after his death.
Perhaps the most challenging aspect of this case is identifying the geographic location of the patient’s exposure to prions. The patient, despite being a US citizen, was born and raised outside of the Americas in 3 countries that were importing UK beef at a time (during 1980–1996) when it was at increased risk for BSE contamination. According to the 2010 US Census, US citizens born outside the Americas constituted <5% of the US population, and not all of this small subgroup would have resided in the United Kingdom or in countries that had imported UK beef during the increased risk period, a residency history common to the 5 previously identified North American vCJD patients (30).
Exportation of BSE-contaminating beef from the United Kingdom during 1980–1996 and/or BSE-infected live cattle during 1980–1990 most likely is the main cause of BSE exposure outside the United Kingdom (4). During 1980–1996, the patient in our report spent >6 years in 2 countries—Kuwait and Russia—to which UK beef was exported. During that time, he also lived <1 year in Lebanon. Although the size of the Kuwaiti population was a few magnitudes smaller, based on UK export data for 1980–1996, the same order of magnitude (≈2.5 × 103 metric tons) of beef was exported to Kuwait and to the Soviet Union/Russia when this patient resided in those 2 countries (4; Her Majesty’s Revenue and Customs, Overseas Trade Statistics, Crops & Trade Branch, Analysis and Evidence Team, unpub. data). These data suggest that the patient’s risk of eating possibly BSE-contaminated UK beef would have been substantially greater during his stay in Kuwait than during his stay in Russia. The risk that he ate such meat was lower in Lebanon than in Russia, primarily because of his relatively short stay in Lebanon during 1980–1996 and the fact that >85% of the 1980–1996 UK beef was imported to Lebanon after he moved to Russia.
This patient lived in the United States for 14 years before vCJD developed. A >14-year incubation period can be consistent with the mean incubation period for vCJD: 11.6–16.7 years, estimated by published models of the vCJD epidemic (31–33). Each of these models predicts a skewed curve toward much longer incubation periods than the mean as the vCJD epidemic wanes over time, similar to other human prion disease outbreaks (31,34,35). In addition, each of these models reflects a strikingly younger distribution of vCJD patients in the United Kingdom and supports the published concept that susceptibility to the prion infection that will cause vCJD peaks among adolescents and declines rapidly with age thereafter (36). Given that this patient did not come to the United States until his late 20s, this age-dependent susceptibility factor favors a conclusion that he was infected before he moved to the United States.
We deemed surgical or transfusion routes of vCJD transmission to or from the patient to be unlikely because the only known surgical procedure he had undergone was a circumcision performed in Kuwait when he was ≈8–10 years of age, and he had no history of receiving or donating blood. A review of records at the Gulf Coast Regional Blood Center and the American Red Cross provided further evidence that he had not been a US blood donor.
Given the markedly declining incidence of vCJD globally, this patient is only the fourth patient worldwide confirmed to have this disease since the beginning of 2012; the other 3 were from the United Kingdom and France (3). This case underscores that the diagnosis of vCJD should not be dismissed if the patient has not resided in a country with a known endemic case of vCJD. Given the several decades’ long potential incubation periods estimated from epidemiologic modeling, the international occurrence of additional vCJD cases can be reasonably anticipated (34,35).
Furthermore, UK surveys of archived appendix tissues indicate an approximate prevalence of asymptomatic vCJD infection of 1 in 2,000 persons born during 1941–1985 (37). Depending on genetic subtype, one may harbor the pathogenic prion and never develop symptoms (3). However, because the agent is transmissible through blood transfusions, organ transplants, and surgical instrumentation, iatrogenic propagation of the disease remains a real possibility (3).
The detection of a definitive case of vCJD in the United States highlights the importance of continuing enhanced national human prion disease surveillance. According to the World Organisation for Animal Health, the BSE status of the 3 countries where the patient reported here resided during the critical exposure period was “undetermined,” suggesting the lack of a systematic BSE surveillance system.
The potential difficulty in making the clinical diagnosis in many patients with vCJD and the delay with which the disease is first suspected raises the concern that vCJD can be missed. The need for neuropathology expertise and advanced neuropathologic techniques is probably an important limiting factor in some parts of the world. The MRI examination, along with the newly developed blood and urine tests, are among the most helpful premortem tests to diagnose vCJD. A postmortem brain and lymphoreticular tissue autopsy examination remains critical to confirm the diagnosis.
Dr. Maheshwari is an assistant professor in the Department of Neurology, Baylor College of Medicine, in Houston, Texas. His focus is on translational research in epilepsy and other neurologic diseases.
References
- Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336:1511–3. DOIPubMedGoogle Scholar
- Barria MA, Ironside JW, Head MW. Exploring the zoonotic potential of animal prion diseases: in vivo and in vitro approaches. Prion. 2014;8:85–91. DOIPubMedGoogle Scholar
- Diack AB, Head MW, McCutcheon S, Boyle A, Knight R, Ironside JW, Variant CJD: 18 years of research and surveillance. Prion. 2014;8:286–95. DOIPubMedGoogle Scholar
- Sanchez-Juan P, Cousens SN, Will RG, van Duijn CM. Source of variant Creutzfeldt-Jakob disease outside United Kingdom. Emerg Infect Dis. 2007;13:1166–9. DOIPubMedGoogle Scholar
- Food and Drug Administration. 2011 Meeting materials, Transmissible Spongiform Encephalopthies Advisory Committee [cited 2014 Oct 1]. http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/TransmissibleSpongiformEncephalopathiesAdvisoryCommittee/ucm261283.htm
- Moda F, Gambetti P, Notari S, Concha-Marambio L, Catania M, Park K-W, Prions in the urine of patients with variant Creutzfeldt-Jakob disease. N Engl J Med. 2014;371:530–9. DOIPubMedGoogle Scholar
- Heath CA, Cooper SA, Murray K, Lowman A, Henry C, MacLeod MA, Diagnosing variant Creutzfeldt-Jakob disease: a retrospective analysis of the first 150 cases in the UK. J Neurol Neurosurg Psychiatry. 2011;82:646–51. DOIPubMedGoogle Scholar
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–5. DOIPubMedGoogle Scholar
- Barash J. Identification of Creutzfeldt-Jakob disease variants. [author reply 1045–6]. Arch Neurol. 2009;66:1045. DOIPubMedGoogle Scholar
- Appleby BS, Appleby KK, Crain BJ, Onyike CU, Wallin MT, Rabins PV. Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch Neurol. 2009;66:208–15 .PubMedGoogle Scholar
- Heath CA, Cooper SA, Murray K, Lowman A, Henry C, MacLeod MA, Validation of diagnostic criteria for variant Creutzfeldt-Jakob disease. Ann Neurol. 2010;67:761–70 .PubMedGoogle Scholar
- World Health Organization Department of Communicable Disease Surveillance and Response. WHO recommended surveillance standards, 1999 [cited 2015 Mar 8]. http://www.who.int/csr/resources/publications/surveillance/whocdscsrisr992.pdf
- Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132:2659–68 . DOIPubMedGoogle Scholar
- Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, Zeidler M, Diagnosing variant Creutzfeldt-Jakob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. AJNR Am J Neuroradiol. 2003;24:1560–9 .PubMedGoogle Scholar
- Ugnon-Café S, Dorey A, Bilheude JM, Streichenberger N, Viennet G, Meyronet D, Rapid screening and confirmatory methods for biochemical diagnosis of human prion disease. J Virol Methods. 2011;175:216–23. DOIPubMedGoogle Scholar
- Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, Thomas DJ, Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet. 1999;353:183–9. DOIPubMedGoogle Scholar
- Lukic A, Mead S, Rudge P, Collinge J. Comment on validation of diagnostic criteria for variant Creutzfeldt-Jakob disease. [author reply 212–3]. Ann Neurol. 2011;69:212. DOIPubMedGoogle Scholar
- Green AJ, Thompson EJ, Stewart GE, Zeidler M, McKenzie JM, MacLeod MA, Use of 14-3-3 and other brain-specific proteins in CSF in the diagnosis of variant Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2001;70:744–8. DOIPubMedGoogle Scholar
- Zeidler M, Stewart GE, Barraclough CR, Bateman DE, Bates D, Burn DJ, New variant Creutzfeldt-Jakob disease: neurological features and diagnostic tests. Lancet. 1997;350:903–7. DOIPubMedGoogle Scholar
- Beck JA, Poulter M, Campbell TA, Adamson G, Uphill JB, Guerreiro R, PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat. 2010;31:E1551–63. DOIPubMedGoogle Scholar
- Jackson GS, Burk-Rafel J, Edgeworth JA, Sicilia A, Abdilahi S, Korteweg J, Population screening for variant Creutzfeldt-Jakob disease using a novel blood test: diagnostic accuracy and feasibility study. JAMA Neurol. 2014;71:421–8. DOIPubMedGoogle Scholar
- Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, Campbell T, Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet. 2011;377:487–93. DOIPubMedGoogle Scholar
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–33. DOIPubMedGoogle Scholar
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br Med Bull. 2003;66:213–39. DOIPubMedGoogle Scholar
- Gambetti P, Cali I, Notari S, Kong Q, Zou W-Q, Surewicz WK. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 2011;121:79–90. DOIPubMedGoogle Scholar
- Sikorska B, Knight R, Ironside JW, Liberski PP. Creutzfeldt-Jakob disease. Adv Exp Med Biol. 2012;724:76–90. DOIPubMedGoogle Scholar
- Mihara M, Sugase S, Konaka K, Sugai F, Sato T, Yamamoto Y, The “pulvinar sign” in a case of paraneoplastic limbic encephalitis associated with non-Hodgkin’s lymphoma. J Neurol Neurosurg Psychiatry. 2005;76:882–4. DOIPubMedGoogle Scholar
- Ryan AM, Ryan J, Wan-Ahmed M, Hardiman O, Farrell MA, McNamara B, Vacuolar leucoencephalopathy and pulvinar sign in association with coeliac disease. BMJ Case Rep. 2009;2009:pii: bcr08.2008.0650. PMID: 20414170
- Schmidt C, Plickert S, Summers D, Zerr I. Pulvinar sign in Wernicke’s encephalopathy. CNS Spectr. 2010;15:215–8 .PubMedGoogle Scholar
- US Census Bureau. The foreign-born population in the United States [cited 2014 Aug 27]. https://www.census.gov/newsroom/pdf/cspan_fb_slides.pdf
- Garske T, Ghani AC. Uncertainty in the tail of the variant Creutzfeldt-Jakob disease epidemic in the UK. PLoS ONE. 2010;5:e15626. DOIPubMedGoogle Scholar
- Ghani AC, Donnelly CA, Ferguson NM, Anderson RM. Updated projections of future vCJD deaths in the UK. BMC Infect Dis. 2003;3:4. DOIPubMedGoogle Scholar
- Valleron AJ, Boelle PY, Will R, Cesbron JY. Estimation of epidemic size and incubation time based on age characteristics of vCJD in the United Kingdom. Science. 2001;294:1726–8. DOIPubMedGoogle Scholar
- Brown P, Brandel J-P, Sato T, Nakamura Y, MacKenzie J, Will RG, Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis. 2012;18:901–7. DOIPubMedGoogle Scholar
- Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Kuru in the 21st century—an acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068–74. DOIPubMedGoogle Scholar
- Boëlle P-Y, Cesbron J-Y, Valleron A-J. Epidemiological evidence of higher susceptibility to vCJD in the young. BMC Infect Dis. 2004;4:26 . DOIPubMedGoogle Scholar
- Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ. 2013;347:f5675. DOIPubMedGoogle Scholar
Figures
Tables
Follow Up
Earning CME Credit
To obtain credit, you should first read the journal article. After reading the article, you should be able to answer the following, related, multiple-choice questions. To complete the questions (with a minimum 75% passing score) and earn continuing medical education (CME) credit, please go to http://www.medscape.org/journal/eid. Credit cannot be obtained for tests completed on paper, although you may use the worksheet below to keep a record of your answers. You must be a registered user on Medscape.org. If you are not registered on Medscape.org, please click on the “Register” link on the right hand side of the website to register. Only one answer is correct for each question. Once you successfully answer all post-test questions you will be able to view and/or print your certificate. For questions regarding the content of this activity, contact the accredited provider, CME@medscape.net. For technical assistance, contact CME@webmd.net. American Medical Association’s Physician’s Recognition Award (AMA PRA) credits are accepted in the US as evidence of participation in CME activities. For further information on this award, please refer to http://www.ama-assn.org/ama/pub/about-ama/awards/ama-physicians-recognition-award.page. The AMA has determined that physicians not licensed in the US who participate in this CME activity are eligible for AMA PRA Category 1 Credits™. Through agreements that the AMA has made with agencies in some countries, AMA PRA credit may be acceptable as evidence of participation in CME activities. If you are not licensed in the US, please complete the questions online, print the certificate and present it to your national medical association for review.
Article Title:
Recent US Case of Variant Creutzfeldt-Jakob Disease—Global Implications
CME Questions
1. You are consulting for a public health department regarding vigilance for emergence of variant Creutzfeldt-Jakob disease (vCJD). According to the case report and review by Maheshwari and colleagues, which of the following statements about the clinical presentation of vCJD is correct?
A. vCJD usually presents with rapidly progressive dementia
B. Development of chorea, ataxia, or other neurologic manifestations supports a diagnosis of sporadic CJD (sCJD) or vCJD
C. Painful sensory complaints are typically reported with sCJD but not vCJD
D. Compared with sCJD, vCJD begins at an older age
2. According to the case report and review by Maheshwari and colleagues, which of the following diagnostic findings is most likely to occur in vCJD?
A. Diffusion restriction and T2 hyperintensity of the cortex and basal ganglia on magnetic resonance imaging
B. Periodic sharp wave complexes on electroencephalogram
C. Elevated 14-3-3/Tau levels in cerebrospinal fluid
D. Detection rate of PrPSc, indicating tonsil biopsy as alternative to brain biopsy
3. Which of the following statements about the global implications of the report by Maheshwari and colleagues of a new US case of vCJD would most likely be correct?
A. The incidence of vCJD is increasing globally
B. vCJD should not be diagnosed if the patient has not lived in a country with a known endemic case of vCJD
C. Iatrogenic transmission of vCJD is unlikely
D. Because the incubation period may be several decades, we can reasonably anticipate the international occurrence of additional vCJD cases
Activity Evaluation
|
1. The activity supported the learning objectives. |
||||
|
Strongly Disagree |
|
|
|
Strongly Agree |
|
1 |
2 |
3 |
4 |
5 |
|
2. The material was organized clearly for learning to occur. |
||||
|
Strongly Disagree |
|
|
|
Strongly Agree |
|
1 |
2 |
3 |
4 |
5 |
|
3. The content learned from this activity will impact my practice. |
||||
|
Strongly Disagree |
|
|
|
Strongly Agree |
|
1 |
2 |
3 |
4 |
5 |
|
4. The activity was presented objectively and free of commercial bias. |
||||
|
Strongly Disagree |
|
|
|
Strongly Agree |
|
1 |
2 |
3 |
4 |
5 |
Related Links
Table of Contents – Volume 21, Number 5—May 2015
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Atul Maheshwari, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA
Top