Volume 23, Number 1—January 2017
Letter
Puumala Virus in Bank Voles, Lithuania
Figure
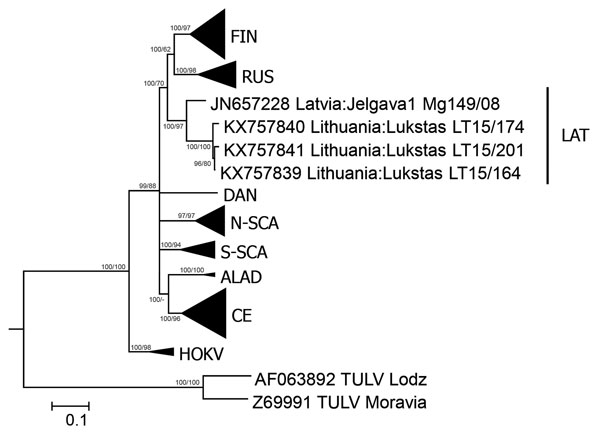
Figure. Phylogenetic tree based on complete nucleocapsid gene sequences of Puumala virus (PUUV) strains from Lithuania (LT), Latvia (Jelgava1), and other PUUV clades. Tula virus (TULV) was used as the outgroup. The tree was generated by Bayesian and maximum-likelihood analysis using MrBayes 3.2.6 (http://mrbayes.sourceforge.net/download.php) and MEGA6 software (http://www.megasoftware.net/). The optimal substitution model was calculated by using jModelTest 2.1.4 (https://code.google.com/p/jmodeltest2). The Bayesian tree was based on transition model 2 with invariant sites and gamma distribution and 4 million generations. For maximum-likelihood analysis, the Kimura 2-parameter model and 1,000 bootstrap replicates were used. Posterior probabilities are indicated before slashes, and bootstrap values are indicated after slashes. Scale bar indicates nucleotide substitutions per site. ALAD, Alpe-Adrian lineage; CE, Central European lineage; DAN, Danish lineage; FIN, Finnish lineage; HOKV, Hokkaido virus; LAT, Latvian lineage; N-SCA, North-Scandinavian lineage; RUS, Russian linage; S-SCA, South-Scandinavian lineage.