Volume 25, Number 1—January 2019
Research
Variable Protease-Sensitive Prionopathy Transmission to Bank Voles
Figure 4
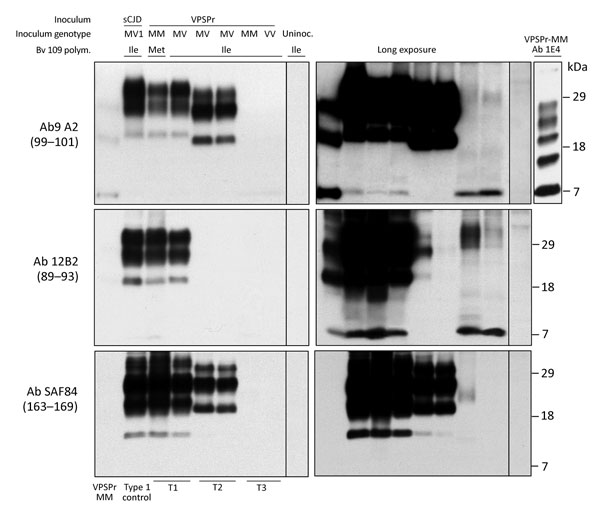
Figure 4. Immunoblot characteristics of protease-resistant, disease-related prion protein (resPrPD) distribution in phenotypes T1–T3 and controls. Regular and long exposures revealed the overall similarity of the 3-band profiles in T1 and T2, but resPrPD profile, including glycoform representation, differed in the 2 phenotypes with all 3 monoclonal antibodies (Ab) used. T1 included a 7-kDa band, not detected in T2, similar to mobility and Ab immunoreactivity of the T3 7-kDa fragment. The T1 profile matched the profile generated in isogenic bank voles inoculated with sCJDMV1 used as human resPrPD type 1 control (ctrl) (lane 2). The T3 profile, visible only after long film exposures, featured a 7-kDa band, but slower migrating bands with variable immunoreactivity were also visible. None of the T1–T3 profiles matched the original VPSPr profile (first lane) although the ≈7-kDa and both 23-kDa and 19-kDa bands were shared with T1 and T2, respectively (compare first with T1 and T2 lanes). The complexity of the native resPrPD profile from VPSPr homogenate is demonstrated by probing with 1E4, a monoclonal Ab to human PrP highly reactive to VPSPr resPrPD (top right panel) (6). Monoclonal Ab 12B2 (middle panels) with high affinity for human resPrPD type 1 confirmed the type 1 characteristics of the resPrPD associated with the T1 phenotype. The small amount of resPrPD type 1 in 1 T2 bank vole probably represents incomplete proteinase K (PK) digestion (lane 5, right panel) (19). Monoclonal Ab SAF84 to the PrP C-terminus, unreactive to human PrP, further underlined the divergence in resPrPD primary structure in T1 and T2 compared with T3. Aside from revealing an additional 13-kDa fragment, strongly detected in T1 and T1 and weakly in T3, SAF84 did not detect the 7-kDa fragment, supporting its internal origin (i.e., cleaved at both N- and C-termini). Uninoculated bank voles were negative for resPrPD. All samples were PK treated. sCJD, sporadic Creutzfeldt-Jakob disease; uninoc., not inoculated; polym., polymorphism; VPSPr, variably protease sensitive prionopathy.
References
- Gambetti P. Creationism and evolutionism in prions. Am J Pathol. 2013;182:623–7. DOIPubMedGoogle Scholar
- Gambetti P, Cali I, Notari S, Kong Q, Zou WQ, Surewicz WK. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 2011;121:79–90. DOIPubMedGoogle Scholar
- Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 2012;11:618–28. DOIPubMedGoogle Scholar
- Parchi P, Strammiello R, Giese A, Kretzschmar H. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathol. 2011;121:91–112. DOIPubMedGoogle Scholar
- Poggiolini I, Saverioni D, Parchi P. Prion protein misfolding, strains, and neurotoxicity: an update from studies on mammalian prions. Int J Cell Biol. 2013;2013:910314.
- Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, et al. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. DOIPubMedGoogle Scholar
- Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I, et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010;68:162–72. DOIPubMedGoogle Scholar
- Notari S, Appleby B, Gambetti P. Variably protease sensitive prionopathy. In: Pocchiari M, Manson J. editors. Handbook of clinical neurology, vol. 153. San Diego: Elsevier; 2018. p. 175–190.
- Collins SJ, Sanchez-Juan P, Masters CL, Klug GM, van Duijn C, Poleggi A, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006;129:2278–87. DOIPubMedGoogle Scholar
- Pirisinu L, Nonno R, Esposito E, Benestad SL, Gambetti P, Agrimi U, et al. Small ruminant nor98 prions share biochemical features with human gerstmann-sträussler-scheinker disease and variably protease-sensitive prionopathy. PLoS One. 2013;8:e66405. DOIPubMedGoogle Scholar
- Rodríguez-Martínez AB, López de Munain A, Ferrer I, Zarranz JJ, Atarés B, Villagra NT, et al. Coexistence of protease sensitive and resistant prion protein in 129VV homozygous sporadic Creutzfeldt-Jakob disease: a case report. J Med Case Reports. 2012;6:348. DOIPubMedGoogle Scholar
- Peden AH, Sarode DP, Mulholland CR, Barria MA, Ritchie DL, Ironside JW, et al. The prion protein protease sensitivity, stability and seeding activity in variably protease sensitive prionopathy brain tissue suggests molecular overlaps with sporadic Creutzfeldt-Jakob disease. Acta Neuropathol Commun. 2014;2:152. DOIPubMedGoogle Scholar
- Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar-Calvo P, et al. Transmission characteristics of variably protease-sensitive prionopathy. Emerg Infect Dis. 2014;20:2006–14. DOIPubMedGoogle Scholar
- Diack AB, Ritchie DL, Peden AH, Brown D, Boyle A, Morabito L, et al. Variably protease-sensitive prionopathy, a unique prion variant with inefficient transmission properties. Emerg Infect Dis. 2014;20:1969–79. DOIPubMedGoogle Scholar
- Nonno R, Angelo Di Bari M, Agrimi U, Pirisinu L. Transmissibility of Gerstmann-Sträussler-Scheinker syndrome in rodent models: New insights into the molecular underpinnings of prion infectivity. Prion. 2016;10:421–33. DOIPubMedGoogle Scholar
- Pirisinu L, Di Bari MA, D’Agostino C, Marcon S, Riccardi G, Poleggi A, et al. Gerstmann-Sträussler-Scheinker disease subtypes efficiently transmit in bank voles as genuine prion diseases. Sci Rep. 2016;6:20443. DOIPubMedGoogle Scholar
- Galeno R, Di Bari MA, Nonno R, Cardone F, Sbriccoli M, Graziano S, et al. Prion strain characterization of a novel subtype of Creutzfeldt-Jakob disease. J Virol. 2017;91:e02390–16. DOIPubMedGoogle Scholar
- Watts JC, Giles K, Patel S, Oehler A, DeArmond SJ, Prusiner SB. Evidence that bank vole PrP is a universal acceptor for prions. PLoS Pathog. 2014;10:e1003990. DOIPubMedGoogle Scholar
- Notari S, Capellari S, Langeveld J, Giese A, Strammiello R, Gambetti P, et al. A refined method for molecular typing reveals that co-occurrence of PrP(Sc) types in Creutzfeldt-Jakob disease is not the rule. Lab Invest. 2007;87:1103–12. DOIPubMedGoogle Scholar
- Zou WQ, Capellari S, Parchi P, Sy MS, Gambetti P, Chen SG. Identification of novel proteinase K-resistant C-terminal fragments of PrP in Creutzfeldt-Jakob disease. J Biol Chem. 2003;278:40429–36. DOIPubMedGoogle Scholar
- Agrimi U, Nonno R, Dell’Omo G, Di Bari MA, Conte M, Chiappini B, et al. Prion protein amino acid determinants of differential susceptibility and molecular feature of prion strains in mice and voles. PLoS Pathog. 2008;4:e1000113. DOIPubMedGoogle Scholar
- Di Bari MA, Chianini F, Vaccari G, Esposito E, Conte M, Eaton SL, et al. The bank vole (Myodes glareolus) as a sensitive bioassay for sheep scrapie. J Gen Virol. 2008;89:2975–85. DOIPubMedGoogle Scholar
- Cosseddu GM, Nonno R, Vaccari G, Bucalossi C, Fernandez-Borges N, Di Bari MA, et al. Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog. 2011;7:e1002370. DOIPubMedGoogle Scholar
- Di Bari MA, Nonno R, Castilla J, D’Agostino C, Pirisinu L, Riccardi G, et al. Chronic wasting disease in bank voles: characterisation of the shortest incubation time model for prion diseases. PLoS Pathog. 2013;9:e1003219. DOIPubMedGoogle Scholar
- Espinosa JC, Nonno R, Di Bari M, Aguilar-Calvo P, Pirisinu L, Fernández-Borges N, et al. PrPC governs susceptibility to prion strains in bank vole, while other host factors modulate strain features. J Virol. 2016;90:10660–9. DOIPubMedGoogle Scholar
- Orrú CD, Groveman BR, Raymond LD, Hughson AG, Nonno R, Zou W, et al. Bank vole prion protein as an apparently universal substrate for RT-QuIC-based detection and discrimination of prion strains. PLoS Pathog. 2015;11:e1004983. DOIPubMedGoogle Scholar
- Fernández-Borges N, Eraña H, Castilla J. Behind the potential evolution towards prion resistant species. Prion. 2018;12:83–7. DOIPubMedGoogle Scholar
- Nonno R, Di Bari MA, Cardone F, Vaccari G, Fazzi P, Dell’Omo G, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog. 2006;2:e12. DOIPubMedGoogle Scholar
- Pirisinu L, Marcon S, Di Bari MA, D’Agostino C, Agrimi U, Nonno R. Biochemical characterization of prion strains in bank voles. Pathogens. 2013;2:446–56. DOIPubMedGoogle Scholar
- Asante EA, Linehan JM, Smidak M, Tomlinson A, Grimshaw A, Jeelani A, et al. Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog. 2013;9:e1003643. DOIPubMedGoogle Scholar
- Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proc Natl Acad Sci U S A. 1998;95:8322–7. DOIPubMedGoogle Scholar
- Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci U S A. 2007;104:4712–7. DOIPubMedGoogle Scholar
- Mays CE, Kim C, Haldiman T, van der Merwe J, Lau A, Yang J, et al. Prion disease tempo determined by host-dependent substrate reduction. J Clin Invest. 2014;124:847–58. DOIPubMedGoogle Scholar
- Mays CE, van der Merwe J, Kim C, Haldiman T, McKenzie D, Safar JG, et al. Prion infectivity plateaus and conversion to symptomatic disease originate from falling precursor levels and increased levels of oligomeric PrPSc species. J Virol. 2015;89:12418–26. DOIPubMedGoogle Scholar
- Weissmann C. Mutation and selection of prions. PLoS Pathog. 2012;8:e1002582. DOIPubMedGoogle Scholar
- Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV. New molecular insight into mechanism of evolution of mammalian synthetic prions. Am J Pathol. 2016;186:1006–14. DOIPubMedGoogle Scholar
- Moreno JA, Telling GC. Insights into mechanisms of transmission and pathogenesis from transgenic mouse models of prion diseases. Methods Mol Biol. 2017;1658:219–52. DOIPubMedGoogle Scholar
- Weissmann C, Li J, Mahal SP, Browning S. Prions on the move. EMBO Rep. 2011;12:1109–17. DOIPubMedGoogle Scholar
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–6. DOIPubMedGoogle Scholar
- Li J, Mahal SP, Demczyk CA, Weissmann C. Mutability of prions. EMBO Rep. 2011;12:1243–50. DOIPubMedGoogle Scholar
- Benedetti D, Fiorini M, Cracco L, Ferrari S, Capucci L, Brocchi E, et al. Molecular characterization of low molecular mass C-terminal fragments in different Creutzfeldt-Jakob disease subtypes. Presented at [Madrid, Spain.]. Prion. 2008;2008:8–10.
1These authors contributed equally to this article.