Volume 25, Number 5—May 2019
Dispatch
Bombali Virus in Mops condylurus Bat, Kenya
Kristian M. Forbes1 , Paul W. Webala, Anne J. Jääskeläinen, Samir Abdurahman, Joseph Ogola, Moses M. Masika, Ilkka Kivistö, Hussein Alburkat, Ilya Plyusnin, Lev Levanov, Essi M. Korhonen, Eili Huhtamo, Dufton Mwaengo, Teemu Smura, Ali Mirazimi, Omu Anzala, Olli Vapalahti, and Tarja Sironen
, Paul W. Webala, Anne J. Jääskeläinen, Samir Abdurahman, Joseph Ogola, Moses M. Masika, Ilkka Kivistö, Hussein Alburkat, Ilya Plyusnin, Lev Levanov, Essi M. Korhonen, Eili Huhtamo, Dufton Mwaengo, Teemu Smura, Ali Mirazimi, Omu Anzala, Olli Vapalahti, and Tarja Sironen
Figure 2
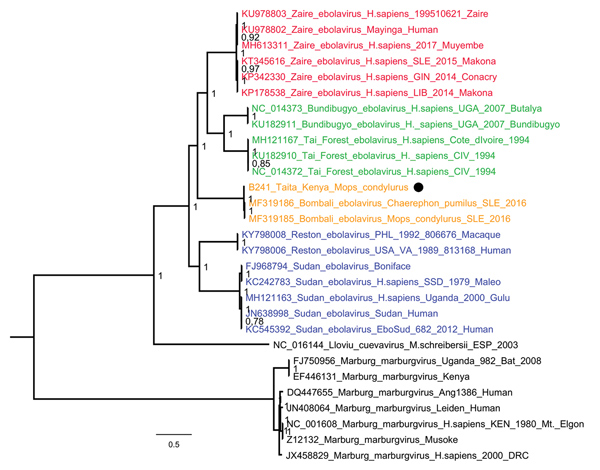
Figure 2. Phylogenetic tree of complete filovirus genomes (18,795–19,115 nt), including Bombali Ebola virus in Sierra Leone and now Kenya (19,026 nt; black dot). Representative sequences were retrieved from the Virus Pathogen Database and Analysis Resource and aligned with a MAFFT online server (http://mafft.cbrc.jp/alignment/software). The tree was built using the Bayesian Markov Chain Monte Carlo method, using a general time reversible model of substitution with gamma-distributed rate variation among sites allowing the presence of invariable sites. Posterior probabilities are shown at the nodes. Scale bar indicates genetic distance.
1Current affiliation: University of Arkansas, Fayetteville, Arkansas, USA.
Page created: April 18, 2019
Page updated: April 18, 2019
Page reviewed: April 18, 2019
The conclusions, findings, and opinions expressed by authors contributing to this journal do not necessarily reflect the official position of the U.S. Department of Health and Human Services, the Public Health Service, the Centers for Disease Control and Prevention, or the authors' affiliated institutions. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.