Volume 27, Number 10—October 2021
Research
New Perspective on the Geographic Distribution and Evolution of Lymphocytic Choriomeningitis Virus, Central Europe
Abstract
Lymphocytic choriomeningitis virus (LCMV) is an Old World mammarenavirus found worldwide because of its association with the house mouse. When LCMV spills over to immunocompetent humans, the virus can cause aseptic meningitis; in immunocompromised persons, systemic infection and death can occur. Central Europe is a strategic location for the study of LCMV evolutionary history and host specificity because of the presence of a hybrid zone (genetic barrier) between 2 house mouse subspecies, Mus musculus musculus and M. musculus domesticus. We report LCMV prevalence in natural mouse populations from a Czech Republic–Germany transect and genomic characterization of 2 new LCMV variants from the Czech Republic. We demonstrate that the main division in the LCMV phylogenetic tree corresponds to mouse host subspecies and, when the virus is found in human hosts, the mouse subspecies found at the spillover location. Therefore, LCMV strains infecting humans can be predicted by the genetic structure of house mice.
Lymphocytic choriomeningitis virus (LCMV) is the prototype of the family Arenaviridae. Its genus Mammarenavirus is associated with rodent-transmitted diseases in humans, including agents of hemorrhagic fevers, such as Lassa virus and Junin virus (1). In immunocompetent persons, LCMV infection is typically asymptomatic but can cause nonspecific febrile illness or aseptic meningitis. However, LCMV infection can cause severe congenital disease, and it has been reported at the origin of 6 clusters of severe or fatal disease among solid organ recipients in the past 20 years (2).
All mammarenaviruses are enveloped ambisense RNA viruses. Their genome (≈11-kb) is composed of 2 segments, each encoding 2 proteins in nonoverlapping open reading frames (ORFs); the large 7.2-kb segment encodes the Z matrix and the large polymerase proteins, and the small 3.4-kb segment encodes the glycoprotein and nucleoprotein (1).
Figure 1
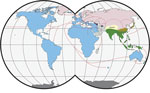
Figure 1. Worldwide distribution of Mus musculus mouse subspecies. Colors indicate subspecies ranges: green and tan, M. musculus castaneus; blue and purple, M. musculus domesticus; pink, ...
The primary host reservoirs of LCMV are house mice (Mus musculus), although the virus has been reported in other rodents, and experimental infections have been described in other mammals, such as rabbits, dogs, and pigs (3–5). The house mouse is a complex of several subspecies. The most widespread subspecies are M. musculus musculus, found from central and northern Europe to the Far East; M. musculus domesticus, which is found in western and southern Europe, northern Africa, the Middle East, and, more recently, in North and South America, southern Africa, Australia, and Oceania because of passive transport with humans; and M. musculus castaneus, which is found in central and southeastern Asia (6,7) (Figure 1). These subspecies are not reproductively isolated, and several regions of secondary contact hybridization exist, including a >2,500 km–long region in Europe extending from Scandinavia to the Black Sea in which M. musculus musculus and M. musculus domesticus mice have contact (Figure 1). In this region, the 2 subspecies form a hybrid zone with a barrier to gene flow between them (6,9,10). Recent studies have shown that such hybrid zones can also act as barriers for the organisms’ pathogens (11–14). Hybrid zones are thus useful natural settings to study the limit of host specificity, which is pivotal to understanding the geographic distribution of pathogens and their potential for spillover. For example, 2 mammarenaviruses, Morogoro virus and Gairo virus, are each confined to 1 of the 2 subtaxa of their host, the Natal mulimammate mouse (Mastomys natalensis), even though the host hybridizes in Tanzania (13,15).
In 2010, Albariño et al. (16) investigated the genetic diversity and distribution of LCMV variants by analyzing 29 genomes. They demonstrated that LCMV is highly diverse and forms 4 distinct lineages (I–IV) but found little correlation of those lineages with time or place of isolation. From their dataset, only 3 strains (Marseille12-2004, Yale-1977, and Michigan-2005) originated from wild mice, but those strains were not assigned to subspecies. Furthermore, the place of isolation is a poor proxy for the origin of spillover to human hosts. For example, focusing on lineage II of Albariño et al., strains M1 and M2 were isolated in Japan in 2005, but came from a wild-derived strain originating from M. musculus musculus mice caught in Illmitz, Austria, in 1985 (17). Likewise, the Dandenong-Yugoslavia LCMV strain (18) was isolated in Australia from a human spillover, but that person returned from the former Yugoslavia before becoming ill and dying. The Bulgaria 1956 strain (19) was isolated from a human spillover, but geographic origin was not mentioned in the original study; a contact of that patient was treated for the same symptoms in a hospital in Vidin, Bulgaria, suggesting spillover origin in northwestern Bulgaria. Finally, the last LCMV strain in lineage II, LE-FRANCE (20), was isolated from a pregnant woman in France (i.e., within M. musculus domesticus mouse territory), but the person worked in a pet store, making strain origin uncertain because other rodent species, especially hamsters, are known to be LCMV carriers (3,4,20). In summary, for 3 of 4 LCMV strains in lineage II, the potential spillover origin is consistent with M. musculus musculus mouse territory despite diverse viral isolation locations. Similarly, in LCMV lineage I, strains were found in laboratory mice, essentially of M. musculus domesticus origin (21); wild mice; or in primate (including human) spillovers in the United States or western Europe, and were thus consistent with M. musculus domesticus mouse origin (22). LCMV lineage IV consists only of strains isolated from woodmice (Apodemus sylvaticus) from Spain. Given these observations, we hypothesized that host specificity could be a better predictor of LCMV genetics than the place or time of LCMV strain isolation.
In this study, we test the hypothesis that LCMV phylogenetic clustering reflects specificity to its host reservoirs by investigating the diversity of LCMV in central Europe across the house mouse hybrid zone (HMHZ). We also update the phylogenetic analysis of LCMV from Albariño et al. (16) by complementing their dataset with LCMV genomes sequenced in the last decade and with our data.
Sampling and Mouse Genotyping
Figure 2
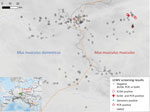
Figure 2. Tested localities for LCMV, Central Europe. The center of the HMHZ is represented by the orange line. The green rectangle n the inset map shows the sampling area, located in...
A total of 748 house mice (410 M. musculus domesticus and 338 M. musculus musculus) from 179 localities (100 for M. musculus domesticus and 79 for M. musculus musculus) were trapped in farms during 2008–2019 across a 145-km by 110-km belt stretching from northeastern Bavaria (Germany) to western Bohemia (Czech Republic), a region in which these mouse subspecies meet and form the HMHZ (23) (Figure 2; Appendix 1 Table 1). Tissue samples were preserved in liquid nitrogen and later stored at −80°C as described in Goüy de Bellocq et al. (24). Mice were identified on the basis of a set of diagnostic markers as in Macholán et al. (23) or on the basis of 1,401 single-nucleotide polymorphism (SNP) markers (25) or 0.62 million SNP markers (26) (Appendix 1 Table 1). Each individual mouse’s hybrid index (HI) was estimated as the proportion of M. musculus musculus alleles. We considered all mice with HIs <0.5 as M. musculus domesticus–like and those with HIs >0.5 as M. musculus musculus–like.
LCMV Serologic and Molecular Screening
We screened 291 blood plasma samples collected from 100 localities during 2008–2011 for LCMV antibodies by using the ELISA kit IM-698 C-EB (XpressBio, https://xpressbio.com). We used 100 μL of 1:50 diluted serum for the reaction according to the manufacturer’s instructions. In addition, we extracted RNA from 616 spleen or salivary gland samples by using RNeasy Mini kit (QIAGEN, https://www.qiagen.com). We reverse-transcribed the RNA samples collected in 2008–2013 by using the Applied Biosystems High-Capacity RNA-to-cDNA Kit (ThermoFisher Scientific, https://www.thermofisher.com) in 10 μL final volume. We screened for LCMV by targeting a 340-nt fragment of the large gene by using primers from Vieth et al. (27), because these primers detected LCMV in a previous study (28). Samples were screened with the Multiplex PCR kit (QIAGEN) in a final volume of 15 μL by using 2 μL of cDNA and following the manufacturer’s instructions. To increase assay sensitivity, we also designed primers for a nested PCR assay on the basis of LCMV sequences available in GenBank and targeting 442 nt in a part of the large gene partially overlapping with the region described previously. We tested 96 samples with both assays and results showed the same number of positive samples. However, the first assay (i.e., Vieth et al. primers) showed higher sensitivity (stronger band in 1.5% agarose gels); therefore, we selected that assay to screen the complete dataset. However, we used the second assay for Sanger sequencing of all positive samples to obtain longer final large fragment (659–665 nt resulting from merging both assay outputs). We screened the 2019 RNA samples with Vieth et al. primers by using Invitrogen SuperScript IV One-Step RT-PCR System (ThermoFisher Scientific) in a final volume of 20 μL and using 3 μL of extracted RNA. We attempted additional amplifications in positive samples to sequence parts of the glycoprotein and nucleoprotein genes (Appendix 2 Table). We purified PCR products and Sanger sequenced in both directions by using Eurofins Genomics (https://eurofinsgenomics.com).
Whole-Genome Sequencing and Assembly of LCMV Viruses
We selected 2 positive samples from localities 10 km apart: sample SK1042 from Kryry, Czech Republic (KRY1) and sample SK1194 from Nepomyšl, Czech Republic (NEPO1), for whole-genome sequencing. We extracted RNA from lung and liver specimens by using the viral enrichment protocol described in Goüy de Bellocq et al. (29). The cDNA synthesis, library preparation, and sequencing (BGI Genomics, https://www.bgi.com) were carried out as described in Goüy de Bellocq et al. (30). After read demultiplexing, quality filtering, and trimming, 48,209,592 paired-end reads were available for SK1042, and 39,228,040 paired-end reads were available for SK1194. We used only 10,000,000 paired-end reads for a de novo assembly by iterative mapping with Geneious Mapper in Geneious 11 (Geneious, https://www.geneious.com). We enriched for LCMV reads in silico by removing all reads that mapped to mouse reference genome GRCm38. The LCMV iterative mapping was seeded with the 340 nt of the large gene obtained by Sanger sequencing and a 74-nt sequence conserved among LCMV strains for the Z gene. For the small segment, we generated 2 small seed reference sequences of ≈150 nt in the glycoprotein and nucleoprotein by first mapping the paired-end reads to LCMV strain Traub (from M. musculus domesticus mice). We confirmed the sequence of the intergenic region of the large segment by Sanger sequencing designing primers in the neighboring coding regions (Appendix 2 Table). After assembly, we ensured the seeding had not influenced the output. Finally, as part of a viral metagenomic study of digestive tract samples taken from mice in the HMHZ (J. Goüy de Belloq, unpub. data), we detected 229-nt and 458-nt contigs that matched via BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) with the large and glycoprotein gene of LCMV and Dandenong virus in a pooled sample of 3 mice coming from Buškovice (BUS2) collected in 2014. We included these 2 sequences in the current study.
Phylogenetic Analyses
LCMV nucleotide sequences were aligned with the sequence coding parts of the nucleoprotein, glycoprotein, and large genes of other strains available in GenBank (Appendix 1 Table 2). We included all strains analyzed in Albariño et al. (16), augmented with 11 recently published genome sequences that included LCMV variants of M. musculus domesticus mouse origin from France, Gabon, and French Guiana (28,31,32). Nucleotide alignment was based on amino acid sequences in Geneious Prime 2019.2 (Geneious) by using the ClustalW algorithm. We used the Bayesian Information Criterion in MEGA X (33) to evaluate models of nucleotide and amino acid substitution. The best-fit model was general time reversible plus invariate sites plus gamma distribution for all 3 genes on the basis of the nucleic acid dataset, Jones-Taylor-Thornton plus invariate sites plus gamma for large and nucleoprotein genes, and Le Gascuel plus gamma for the glycoprotein gene for the amino acid dataset. We performed phylogenetic analyses on nucleic acid and amino acid sequences by using Bayesian inference using MrBayes version 3.2.7 (34). For the amino acid sequence dataset, we only included LCMV sequences in which a large proportion of the coding genes were sequenced. We conducted default priors for all parameters and 2 independent runs with 10 million generations per run and sampled trees and parameters every 500 generations, discarding the first 25% of sampled trees as burn-in. We used Bayesian posterior probabilities (PP) to assess node support and the complete genome of Lunk virus from African Mus minutoides as an outgroup. We prepared tree figures in FigTree version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree).
Rodent Sampling and LCMV Detection
We found 7 positive samples among the 291 samples (160 M. musculus domesticus and 131 M. musculus musculus) tested with ELISA for a prevalence of 2.4%. A total of 6 positives were revealed in M. musculus musculus mouse territory: 3 in Buškovice (BUS2, 2008), 1 in Nepomyšl (NEPO1, 2009), 1 in Kryry (KRY1, 2009), and 1 in Žihle (2010). All these specimens had HI >0.96, indicating almost pure M. musculus musculus. The single positive specimen from M. musculus domesticus territory mouse (HI = 0.20) was captured in locality Starý Rybník Vepřín (SRYV) (2009), 3.7 km from the center of the HMHZ. By using the molecular LCMV screening, we found 5 positive samples out of 616 analyzed (prevalence = 0.8%) (Table). All were from M. musculus musculus mouse territory with HI >0.97: 4 specimens from NEPO1 (2008 and 2009) and 1 specimen from KRY1 (2009). One specimen from NEPO1 (SK1042; HI = 0.98) was found positive by both serologic and molecular screening approaches. All the positive localities confirmed genetically were located within a 12-km2 area. The detection of RNA-positive samples in 2009 and 2010 in NEPO1 and the repeated finding of positive specimens in BUS2 by ELISA in 2008 and 2014 (J. Goüy de Belloq, unpub. data) suggest that LCMV is locally endemic in M. musculus musculus mouse territory, persisting within farms over several years.
Characterization of the Full Genomes of LCMV from the Czech Republic
We obtained LCMV whole-genome sequences from 2 mice samples. Because the partial large sequences of the 4 samples from NEPO1 were identical, we characterized the genome of only 1 LCMV sample (SK1194). The other sample (SK1042) was from the locality KRY1, 10 km from NEPO1. At the 3′ end of the large segment of the variant SK1194, ≈19 noncoding nucleotides were missing. Each of the 2 segments showed the 2 ORFs typical for mammarenavirus separated by typical stem-loop structures. The complete large segment was 7232 nt (for strain SK1042) and contained 2 ORFs: the Z ORF (270 nt) encodes a 90-aa zinc-finger protein, whereas the large ORF (6627 nt) encodes a 2209-aa RNA-dependent RNA polymerase. The complete small segment was 3380 nt for SK1194 and 3381 nt for SK1042 and contained 2 ORFs: the glycoprotein ORF (1494 nt) coding for a 498-aa glycoprotein precursor and the nucleoprotein ORF (1674 nt) encoding a 558-aa nucleoprotein. The pairwise nucleotide divergence between the 2 variants was 8.4% for the small segment and 9.6% for the large segment.
Phylogenetic Analysis
Figure 3

Figure 3. Phylogenetic analysis performed on nucleic acid sequences of large gene of lymphocytic choriomeningitis virus (LCMV) sequences using Bayesian inference. Bayesian posterior probabilities were used to assess node support. Lunk virus...
We analyzed the large, glycoprotein, and nucleoprotein genes separately and highlighted the position of the new LCMV variants found in M. musculus musculus mice from the Czech Republic and of the variants known to have been isolated from wild M. musculus domesticus mice. For the large nucleotide tree (Figure 3), the topology of the phylogeny is similar to that of Albariño et al. (16); 2 lineages are highly supported (PP = 0.99): I (harboring 24 LCMV strains) and II (10 LCMV strains). Lineage III, which consisted of a single sequence isolated from a human in Georgia (USA), has a highly supported basal position (PP = 1). All sequences from Czech Republic M. musculus musculus mice form a sister clade to Dandenong virus within lineage II, the lineage we hypothesized to originate from M. musculus musculus mice. The 3 variants from M. musculus domesticus mice are found scattered within the highly supported lineage I (PP = 0.99). The internal topology of lineage I is not resolved and consists of sequences originating from various hosts and regions. Among these clades, a subset was isolated from primate spillovers, likely from local mice in the United States, Spain, France, and Germany (i.e., within M. musculus domesticus mouse territory). The other strains that do not overlap with the range of M. musculus domesticus mice have either been isolated from cell culture (strains from Japan or Slovakia) or ticks (strains from China). In the phylogenetic tree constructed on the basis of large amino acid sequences, the position of lineage III is again well supported but is basal to lineages I and II, both with highly supported monophyly (PP = 1) (Appendix 2 Figure, panel A).
Figure 4

Figure 4. Phylogenetic analysis performed on nucleic acid sequences of glycoprotein gene of lymphocytic choriomeningitis virus (LCMV) sequences using Bayesian inference. Bayesian posterior probabilities were used to assess node support. Lunk virus...
Figure 5

Figure 5. Phylogenetic analysis performed on nucleic acid sequences of nucleoprotein gene of lymphocytic choriomeningitis virus (LCMV) sequences using Bayesian inference. Bayesian posterior probabilities were used to assess node support. Lunk virus...
The phylogenetic position of the sequences from Czech Republic M. musculus musculus and wild M. musculus domesticus mice in our nucleoprotein and glycoprotein gene trees corresponds to that in the large gene tree. An additional clade, clade IV, is composed of strains isolated from the woodmouse (Apodemus sylvaticus). All 4 glycoprotein lineages based on amino acid sequences were highly supported (PP = 1) (Appendix 2 Figure, panel B), whereas the phylogenetic signal at the nucleotide level seems to be compromised by homoplasy, resulting in trichotomy between lineages I, II, and III (Figure 4). A similar pattern can be seen in the phylogenetic trees based on the nucleoprotein gene but with low support. Phylogenetic relationships between lineages are not resolved, demonstrating differences with regard to the type of data. The basal position of lineage IV (woodmouse) to other lineages is well supported (PP = 1) on the basis of amino acid sequences (Appendix 2 Figure, panel C). By contrast, nucleotide sequences show lineage IV as sister group to lineage I (PP = 0.92) and lineage III clustering with lineage II (PP = 1), whereas the strain from Bulgaria is basal to all other ingroup lineages (PP = 1), suggesting that homoplasy at the nucleic acid level affects the phylogenetic signal (Figure 5).
We found LCMV at low prevalence in wild mice in central Europe, and all genetically confirmed cases clustered within a small geographic region in the M. musculus musculus mouse side of the HMHZ. This low prevalence prevents direct inference of the zone as a barrier to LCMV exchange between the mouse subspecies in nature. However, our phylogenetic analyses, which included new LCMV variants from the Czech Republic, 3 variants sequenced from wild M. musculus domesticus mice, other LCMV variants sequenced during the last decade, and supplemented with published data, support the hypothesis that LCMV lineage I harbors viruses originating from M. musculus domesticus mice and lineage II includes viruses primarily found in M. musculus musculus mice.
The low prevalence of LCMV observed in central Europe is not uncommon. In wild mice, this prevalence has been shown to be variable, ranging from 0 to 25% (2), but most studies have reported low prevalence and patchy distribution. For example, Ackermann et al. (34) found an overall prevalence of 3% in wild mice from Germany, with 65 LCMV-positive specimens from 44 localities, but despite extensive sampling efforts in Bavaria as a whole (380 mouse samples over 70,000 km2), no LCMV-positive mice were found there (35). We also failed to detect any positive LCMV samples in Bavaria (M. musculus domesticus mouse region). The low prevalence of LCMV is comparable to other mammarenaviruses (e.g., Gairo virus and Morogoro virus in Mastomys natalensis mice in Tanzania) (13,15).
We reported LCMV infection in Buškovice in 2008 and 2014; however, we were unable to demonstrate genetic turnover during that period. Commensal mouse populations are usually structured to local subpopulations or demes, with a dispersal scale of ≈1 km2 (36,37). Because LCMV can spread both horizontally and vertically, maintenance of the virus within a deme over several years seems plausible. Whether LCMV variants are still present in the 12 km2 area is not certain. If so, targeted rodent control measures could feasibly decrease or eliminate LCMV risk for humans in this geographic area.
Albariño et al. (16) described 4 main LCMV lineages. Our results suggest that >3 of these lineages correspond to different host subspecies: lineage I to M. musculus domesticus, lineage II to M. musculus musculus, and lineage IV to Apodemus sylvaticus. We make no claim regarding the origin of lineage III, a single isolate from a human in Georgia (USA) (i.e., theoretically M. musculus domesticus mouse territory). We suggest more highly divergent lineages are likely to be discovered corresponding to rodent species, subspecies, and cryptic taxa. A new LCMV strain was recently reported from human serum in southern Iraq (38), but its phylogenetic position cannot be resolved; only a short fragment of the large gene (395 nt) is available in GenBank. This new LCMV strain is likely to cluster in clade I because M. musculus domesticus is the expected house mouse subspecies in southern Iraq (39–41). Uncertainty persists with respect to 4 LCMV strains clustered within lineage I of expected M. musculus domesticus mouse origin; JX14, JX4, and JX31 were isolated from ticks in 2015 from a coastal area in Jinxin, Jilin Province, northeastern China, and strain OQ28 was sequenced in 1990 from a wild mouse (M. musculus) captured in Osaka, Japan (42,43). In both regions, mice of subspecies other than M. musculus domesticus were reported. M. musculus musculus mice occur in northern China (44), whereas in Japan, mice are generally identified as M. musculus castaneus or M. musculus molossinus (45). However, the M. musculus domesticus mouse is known to be a successful invasive species because of ancient and recent human mobility, and its introduction to new areas is regularly reported, particularly in port cities, coastal areas, and islands (6). This expansion might explain the presence of M. musculus domesticus LCMV strains in Osaka and Jinxin, both coastal areas.
LCMV can take a severe toll on human health, particularly in immunosuppressed persons. Cases of death after organ transplant have been reported involving strains from both lineages I and II (3,18,46). Takagi et al. (41) showed that 3 LCMV strains—OQ28, WE, and BRC—differ in pathogenicity in mice, concluding that strains OQ28 and BRC were genetically classified within the same cluster but exhibited very different pathogenicity. In this study, we demonstrate that the OQ28 strain clusters to M. musculus domesticus lineage I and the BRC strain clusters to M. musculus musculus lineage II; thus, we propose the 2 lineages have different host origins. From this perspective, the differences observed in strain pathogenicity by Takagi et al. (41) seem less surprising. Nevertheless, the variation of pathogenicity of LCMV strains corresponding to other host taxa is currently unknown.
In conclusion, our results suggest that the evolutionary diversity of LCMV might reflect rodent expansion history. When a human LCMV infection is diagnosed, sampling efforts should be applied to any synanthropic rodents. This effort could help clarify LCMV evolutionary history and elucidate whether different lineages differ in their spillover ability.
Dr. Fornůsková is a research assistant at the Institute of Vertebrate Biology, Czech Academy of Sciences, Czech Republic. Her research is focused on host-pathogen interactions with special attention on small mammals as reservoir hosts.
Acknowledgments
We thank Stuart J.E. Baird for English proofreading and final improvement of the article, and many colleagues and local farmers for their help trapping mice. We are grateful to Mark Blaxter, Konrad Lohse, Kamil Jaroň, and Jordan Asworth for the warm welcoming of A. Fornuskova at the University of Edinburgh during her internship and introducing her to the world of genomic analyses. We are indebted to Anne Lavergne for providing us genetic information of positive mouse samples from French Guiana and to Vladislav Vergilov for his assistance with translating a Bulgarian article and hence with locating the geographic origin of LCMV-Bulgaria.
This work was supported by the Czech Science Foundation (grant no. 16-20049S) and the Program of the Czech Academy of Science to support the international cooperation of early-stage researchers (grant no. MSM200931901). The European Society for Evolutionary Biology awarded A. Fornuskova the Godfrey Hewitt Mobility Award, which enabled her to combine scientific work and family life during her stay in the United Kingdom. Computational resources were supplied by the project e-Infrastruktura CZ (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures. Samples were collected with the support of the project 16-23773S of the Czech Science Foundation and the project 39022/2018-1 of the Ministry of Education, Youth, and Sports.
References
- Radoshitzky SR, Buchmeier MJ, Charrel RN, Clegg JCS, Gonzalez JJ, Günther S, et al.; Ictv Report Consortium. ICTV virus taxonomy profile: Arenaviridae. J Gen Virol. 2019;100:1200–1. DOIPubMedGoogle Scholar
- Childs JE, Klein SL, Glass GE. A case study of two rodent-borne viruses: not always the same old suspects. Front Ecol Evol. 2019;7:35. DOIGoogle Scholar
- Amman BR, Pavlin BI, Albariño CG, Comer JA, Erickson BR, Oliver JB, et al. Pet rodents and fatal lymphocytic choriomeningitis in transplant patients. Emerg Infect Dis. 2007;13:719–25. DOIPubMedGoogle Scholar
- Parker JC, Igel HJ, Reynolds RK, Lewis AM Jr, Rowe WP. Lymphocytic choriomeningitis virus infection in fetal, newborn, and young adult Syrian hamsters (Mesocricetus auratus). Infect Immun. 1976;13:967–81. DOIPubMedGoogle Scholar
- Skinner HH, Knight EH. The potential role of Syrian hamsters and other small animals as reservoirs of lymphocytic choriomeningitis virus. J Small Anim Pract. 1979;20:145–61. DOIPubMedGoogle Scholar
- Boursot P, Auffray JC, Britton-Davidian J, Bonhomme F. The evolution of house mice. Annu Rev Ecol Syst. 1993;24:119–52. DOIGoogle Scholar
- Didion JP, de Villena FP-M. Deconstructing Mus gemischus: advances in understanding ancestry, structure, and variation in the genome of the laboratory mouse. Mamm Genome. 2013;24:1–20. DOIPubMedGoogle Scholar
- Phifer-Rixey M, Nachman MW. Insights into mammalian biology from the wild house mouse Mus musculus. eLife. 2015;4:
e05959 . DOIPubMedGoogle Scholar - Baird S, Macholán M. What can the Mus musculus musculus/M. m. domesticus hybrid zone tell us about speciation. In: Macholán M, Baird S, Munclinger P, Piálek J, editors. Evolution of the house mouse. Cambridge: Cambridge University Press; 2012. p. 334–72.
- Ďureje L, Macholán M, Baird SJE, Piálek J. The mouse hybrid zone in Central Europe: from morphology to molecules. Folia Zool (Brno). 2012;61:308–18. DOIGoogle Scholar
- Goüy de Bellocq J, Wasimuddin , Ribas A, Bryja J, Piálek J, Baird SJE. Holobiont suture zones: Parasite evidence across the European house mouse hybrid zone. Mol Ecol. 2018;27:5214–27. DOIPubMedGoogle Scholar
- Čížková D, Baird SJE, Těšíková J, Voigt S, Ľudovít Ď, Piálek J, et al. Host subspecific viral strains in European house mice: Murine cytomegalovirus in the Eastern (Mus musculus musculus) and Western house mouse (Mus musculus domesticus). Virology. 2018;521:92–8. DOIPubMedGoogle Scholar
- Gryseels S, Baird SJE, Borremans B, Makundi R, Leirs H, Goüy de Bellocq J. When viruses don’t go viral: the importance of host phylogeographic structure in the spatial spread of Arenaviruses. PLoS Pathog. 2017;13:
e1006073 . DOIPubMedGoogle Scholar - Kváč M, McEvoy J, Loudová M, Stenger B, Sak B, Květoňová D, et al. Coevolution of Cryptosporidium tyzzeri and the house mouse (Mus musculus). Int J Parasitol. 2013;43:805–17. DOIPubMedGoogle Scholar
- Cuypers LN, Baird SJE, Hánová A, Locus T, Katakweba AS, Gryseels S, et al. Three arenaviruses in three subspecific natal multimammate mouse taxa in Tanzania: same host specificity, but different spatial genetic structure? Virus Evol. 2020;6:a039; Epub ahead of print. DOIPubMedGoogle Scholar
- Albariño CG, Palacios G, Khristova ML, Erickson BR, Carroll SA, Comer JA, et al. High diversity and ancient common ancestry of lymphocytic choriomeningitis virus. Emerg Infect Dis. 2010;16:1093–100. DOIPubMedGoogle Scholar
- Ike F, Bourgade F, Ohsawa K, Sato H, Morikawa S, Saijo M, et al. Lymphocytic choriomeningitis infection undetected by dirty-bedding sentinel monitoring and revealed after embryo transfer of an inbred strain derived from wild mice. Comp Med. 2007;57:272–81.PubMedGoogle Scholar
- Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358:991–8. DOIPubMedGoogle Scholar
- Bozhinov S, Shindarov L, Makedonska D. [Clinical and virologic examination of lymphocytic choriomeningitis] [in Bulgarian]. Suvr Med (Sofiia). 1956;7:49–59.PubMedGoogle Scholar
- Meritet JF, Krivine A, Lewin F, Poissonnier MH, Poizat R, Loget P, et al. A case of congenital lymphocytic choriomeningitis virus (LCMV) infection revealed by hydrops fetalis. Prenat Diagn. 2009;29:626–7. DOIPubMedGoogle Scholar
- Lilue J, Doran AG, Fiddes IT, Abrudan M, Armstrong J, Bennett R, et al. Sixteen diverse laboratory mouse reference genomes define strain-specific haplotypes and novel functional loci. Nat Genet. 2018;50:1574–83. DOIPubMedGoogle Scholar
- Tichy H, Zaleska-Rutczynska Z, O’Huigin C, Figueroa F, Klein J. Origin of the North American house mouse. Folia Biol (Praha). 1994;40:483–96.PubMedGoogle Scholar
- Macholán M, Baird SJE, Dufková P, Munclinger P, Bímová BV, Piálek J. Assessing multilocus introgression patterns: a case study on the mouse X chromosome in central Europe. Evolution. 2011;65:1428–46. DOIPubMedGoogle Scholar
- Goüy de Bellocq J, Baird SJE, Albrechtová J, Sobeková K, Piálek J. Murine cytomegalovirus is not restricted to the house mouse Mus musculus domesticus: prevalence and genetic diversity in the European house mouse hybrid zone. J Virol. 2015;89:406–14. DOIPubMedGoogle Scholar
- Wang L, Luzynski K, Pool JE, Janoušek V, Dufková P, Vyskočilová MM, et al. Measures of linkage disequilibrium among neighbouring SNPs indicate asymmetries across the house mouse hybrid zone. Mol Ecol. 2011;20:2985–3000. DOIPubMedGoogle Scholar
- Yang H, Wang JR, Didion JP, Buus RJ, Bell TA, Welsh CE, et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet. 2011;43:648–55. DOIPubMedGoogle Scholar
- Vieth S, Drosten C, Lenz O, Vincent M, Omilabu S, Hass M, et al. RT-PCR assay for detection of Lassa virus and related Old World arenaviruses targeting the L gene. Trans R Soc Trop Med Hyg. 2007;101:1253–64. DOIPubMedGoogle Scholar
- Yama IN, Cazaux B, Britton-Davidian J, Moureau G, Thirion L, de Lamballerie X, et al. Isolation and characterization of a new strain of lymphocytic choriomeningitis virus from rodents in southwestern France. Vector Borne Zoonotic Dis. 2012;12:893–903. DOIPubMedGoogle Scholar
- Goüy de Bellocq J, Těšíková J, Meheretu Y, Čížková D, Bryjová A, Leirs H, et al. Complete genome characterisation and phylogenetic position of Tigray hantavirus from the Ethiopian white-footed mouse, Stenocephalemys albipes. Infect Genet Evol. 2016;45:242–5. DOIPubMedGoogle Scholar
- Goüy de Bellocq J, Bryjová A, Martynov AA, Lavrenchenko LA. Dhati Welel virus, the missing mammarenavirus of the widespread Mastomys natalensis. J Vertebr Biol. 2020;69:20018.1–11.
- Lavergne A, de Thoisy B, Tirera S, Donato D, Bouchier C, Catzeflis F, et al. Identification of lymphocytic choriomeningitis mammarenavirus in house mouse (Mus musculus, Rodentia) in French Guiana. Infect Genet Evol. 2016;37:225–30. DOIPubMedGoogle Scholar
- N′Dilimabaka N, Berthet N, Rougeron V, Mangombi JB, Durand P, Maganga GD, et al. Evidence of lymphocytic choriomeningitis virus (LCMV) in domestic mice in Gabon: risk of emergence of LCMV encephalitis in Central Africa. J Virol. 2014;89:1456–60.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35:1547–9. DOIPubMedGoogle Scholar
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42. DOIPubMedGoogle Scholar
- Ackermann R, Bloedhorn H, Küpper B, Winkens I, Scheid W. [Spread of the lymphocytic choriomeningitis virus among West German mice. I. Investigations mostly on domestic mice (Mus musculus)] [in German]. Zentralbl Bakteriol Orig. 1964;194:407–30.PubMedGoogle Scholar
- Macholán M, Munclinger P, Sugerková M, Dufková P, Bímová B, Bozíková E, et al. Genetic analysis of autosomal and X-linked markers across a mouse hybrid zone. Evolution. 2007;61:746–71. DOIPubMedGoogle Scholar
- Pocock MJO, Hauffe HC, Searle JB. Dispersal in house mice. Biol J Linn Soc Lond. 2005;84:565–83. DOIGoogle Scholar
- Alburkat H, Jääskeläinen AJ, Barakat AM, Hasony HJ, Sironen T, Al-Hello H, et al. Lymphocytic choriomeningitis virus infection, Southern Iraq. Emerg Infect Dis. 2020;26:3002–6. DOIPubMedGoogle Scholar
- Carleton M, Musser GG. Order Rodentia. In: Wilson DE, Reeder DM, editors. Mammal species of the world: a taxonomic and geographic reference. 3rd ed. Baltimore: The Johns Hopkins University Press; 2005. p. 745–52.
- Hardouin EA, Orth A, Teschke M, Darvish J, Tautz D, Bonhomme F. Eurasian house mouse (Mus musculus L.) differentiation at microsatellite loci identifies the Iranian plateau as a phylogeographic hotspot. BMC Evol Biol. 2015;15:26. DOIPubMedGoogle Scholar
- Shad H, Darvish J, Rastegar Pouyani E, Mahmoudi A. Subspecies differentiation of the house mouse Mus musculus Linnaeus, 1758 in the center and east of the Iranian plateau and Afghanistan. Mammalia. 2016;81:147–68.
- Takagi T, Ohsawa M, Morita C, Sato H, Ohsawa K. Genomic analysis and pathogenic characteristics of lymphocytic choriomeningitis virus strains isolated in Japan. Comp Med. 2012;62:185–92.PubMedGoogle Scholar
- Zhang L, Li S, Huang S-J, Wang Z-D, Wei F, Feng X-M, et al. Isolation and genomic characterization of lymphocytic choriomeningitis virus in ticks from northeastern China. Transbound Emerg Dis. 2018;65:1733–9. DOIPubMedGoogle Scholar
- Jing M, Yu H-T, Bi X, Lai Y-C, Jiang W, Huang L. Phylogeography of Chinese house mice (Mus musculus musculus/castaneus): distribution, routes of colonization and geographic regions of hybridization. Mol Ecol. 2014;23:4387–405. DOIPubMedGoogle Scholar
- Yonekawa H, Moriwaki K, Gotoh O, Miyashita N, Matsushima Y, Shi LM, et al. Hybrid origin of Japanese mice “Mus musculus molossinus”: evidence from restriction analysis of mitochondrial DNA. Mol Biol Evol. 1988;5:63–78.PubMedGoogle Scholar
- Fischer SA, Graham MB, Kuehnert MJ, Kotton CN, Srinivasan A, Marty FM, et al.; LCMV in Transplant Recipients Investigation Team. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N Engl J Med. 2006;354:2235–49. DOIPubMedGoogle Scholar
Figures
Cite This ArticleOriginal Publication Date: August 27, 2021
Table of Contents – Volume 27, Number 10—October 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Alena Fornůsková, Institute of Vertebrate Biology of the Czech Academy of Sciences, Květná 170/8, 603 65 Brno, Czech Republic
Top