Volume 28, Number 3—March 2022
Synopsis
Airborne Transmission of SARS-CoV-2 Delta Variant within Tightly Monitored Isolation Facility, New Zealand (Aotearoa)
Figure 3
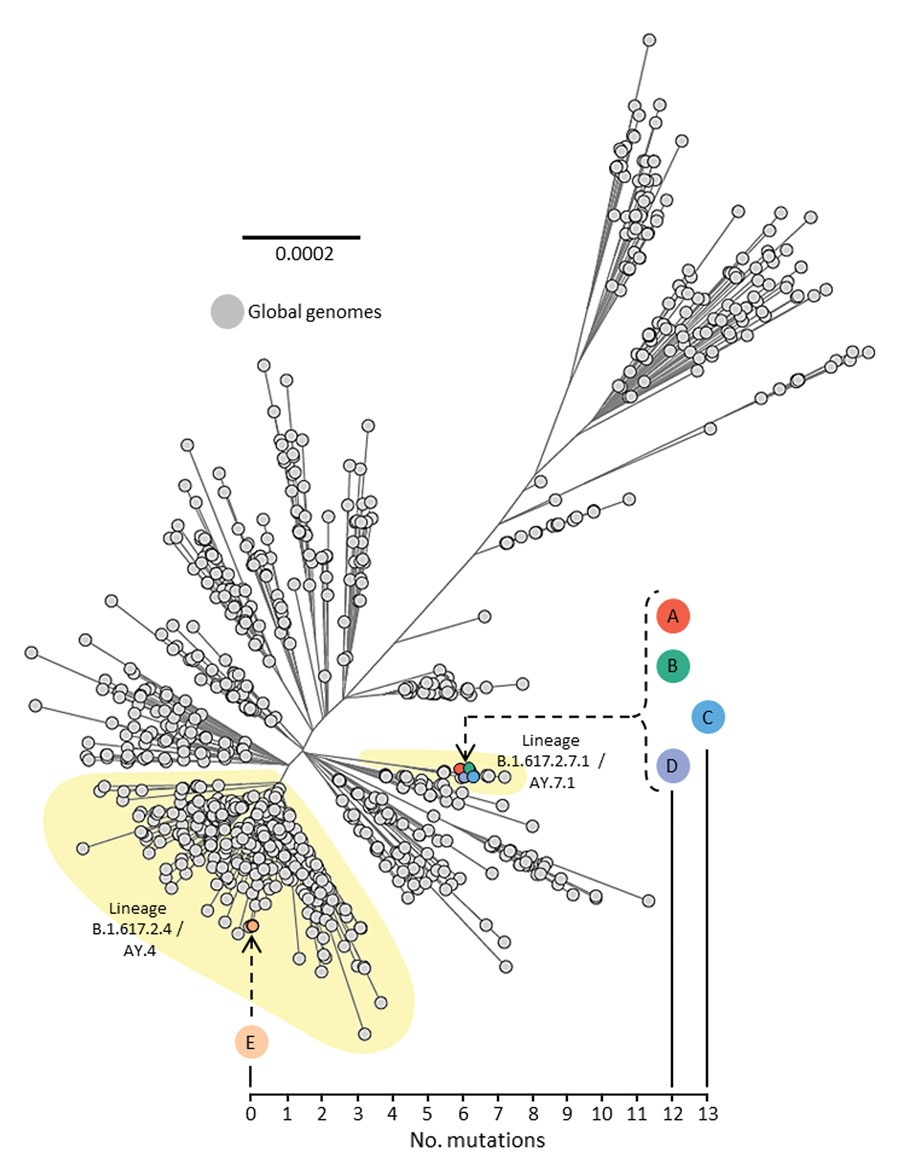
Figure 3. Unrooted maximum-likelihood phylogenetic tree of genomes from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) isolated from persons A, B, C, D, and E, implicated in airborne transmission of SARS-CoV-2 Delta variant between separate nonadjacent rooms within a tightly monitored managed isolation facility, New Zealand, set among a background of other lineage B.1.617.2 (Delta variant) genomes sampled from around the world during July 1–14, 2021. Colored circles indicate persons A–E. Person F is not included because they were not infected with SARS-CoV-2 during the timeframe of this investigation. Upper left phylogenetic scale bar indicates number of nucleotide substitutions per site. Lower right scale shows number of mutations (single nucleotide polymorphisms) difference between viral sequences isolated from persons A, B, C, D, and E.