Volume 28, Number 9—September 2022
Research Letter
Molecular Epidemiology of Blastomyces gilchristii Clusters, Minnesota, USA
Abstract
We characterized 2 clusters of blastomycosis cases in Minnesota, USA, using whole-genome sequencing and single-nucleotide polymorphism analyses. Blastomyces gilchristii was confirmed as the cause of infection. Genomic analyses corresponded with epidemiologic findings for cases of B. gilchristii infections, demonstrating the utility of genomic methods for future blastomycosis outbreak investigations.
Three pathogenic Blastomyces species, B. dermatitidis, B. gilchristii, and B. helicus, have been identified in North America. In the United States, B. dermatitidis has been found throughout areas surrounding the Great Lakes, the Ohio and Mississippi River valleys, and the St. Lawrence River (1). In contrast, B. gilchristii has a smaller geographic range in Canada and the northern United States (2), and B. helicus has been found in the northwestern United States (3). No differences in clinical manifestations have been reported among these Blastomyces species.
In the United States, previous case reports have linked blastomycosis infections to outdoor activities, especially those involving moist soil and proximity to waterways (4,5). One of the largest reported outbreaks of blastomycosis occurred in 2015 among persons who had recreated along the Little Wolf River in Wisconsin (6). In Minnesota, blastomycosis is a reportable disease; epidemiologists at the Minnesota Department of Health (MDH) routinely collect demographic and clinical information for blastomycosis cases and attempt interviews to characterize illness and exposure history. The MDH Public Health Laboratory provides fungal identification services and stores isolates submitted by clinical laboratories.
Although whole-genome sequencing has been used to investigate outbreaks involving various fungal pathogens, such as Candida auris and Coccidioides spp. (7,8), this molecular technology has not been used to investigate Blastomyces spp. outbreaks in the United States. We performed whole-genome sequencing to determine the genetic diversity and phylogenetic relationships of 2 familial clusters of B. gilchristii infections identified in Minnesota.
In August 2020, five cases of blastomycosis were identified as cluster A, which comprised a family of 2 White Hispanic parents and 3 children (Table). Four of the 5 patients were hospitalized, of which 3 had sputum cultures that were positive for Blastomyces sp. All 5 patients recovered from illness. The mother reported that the family had visited rivers in St. Croix County, Wisconsin, numerous times during the summer. No other likely exposure locations or activities were reported.
In addition, 2 cases of blastomycosis were identified in White non-Hispanic sisters. Only 1 sister was hospitalized and had a positive culture for Blastomyces sp. from a bronchoalveolar lavage specimen. MDH learned that their father had blastomycosis in 2014, which was attributed to B. dermatitidis (9). The 2 patients with isolates (1 sister and the father) were classified as cluster B (Table). The family owned a cabin in Hubbard County, Minnesota, which is highly endemic for blastomycosis and was likely the exposure location for the three cases. All 3 patients recovered from illness.
Blastomyces identification is routinely performed by MDH only at the genus level. Therefore, the Centers for Disease Control and Prevention (CDC) determined the species in 4 isolates from the 2 blastomycosis clusters and performed Illumina (https://www.illumina.com) short-read sequencing (National Center for Biotechnology Information BioProject accession no. PRJNA786864). To investigate genetic diversity between strains, we performed whole-genome single-nucleotide polymorphism (SNP) analysis using the MycoSNP version 0.19 analytical workflow (https://github.com/CDCgov/mycosnp). We used publicly available sequences from B. dermatitidis isolates (NCBI run nos. SRR11849827, SRR11849828, SRR11849829) for comparison and genome assembly data for B. gilchristii strain SLH14081 from GenBank (accession no. GCA_000003855.2) as a reference. We constructed a neighbor-joining tree showing SNP differences and maximum-likelihood tree showing bootstrap values using MEGA software version 7.0, (https://www.megasoftware.net) and FastTree 2 (10).
Figure
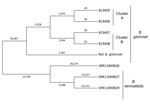
Figure. Genetic relationships and molecular epidemiology of Blastomyces gilchristii clusters, Minnesota, USA. We performed whole-genome sequencing of isolates from 4 patients in Minnesota who had Blastomyces gilchristiiinfections...
All the isolates were B. gilchristii rather than B. dermatitidis. Phylogenetic tree analysis showed B. dermatitidis and B. gilchristii grouped into distinct clades, which were separated by 52,431 SNPs (Figure). Sequences from all 4 B. gilchristii isolates clustered with the reference genome SLH14081 and were separated by a minimum of 11,695 SNPs. Each familial cluster formed a subclade within the B. gilchristii clade; the subclades were separated by 5,214 SNPs. In cluster A, where all family members were infected at the same time and location, we found 63 SNPs separated the 2 cases. In cluster B, where exposures occurred in the same location but infections were 6 years apart, the cases differed by 120 SNPs (Figure).
Both B. dermatitidis and B. gilchristii have been reported in Minnesota (2). We used whole-genome sequencing and SNP analysis to evaluate clusters of blastomycosis infections caused by B. gilchristii in Minnesota. The genomic data showed that cases within cluster A or B were closely related genetically, whereas clusters A and B were genetically distinct. B. gilchristii is likely responsible for a higher proportion of blastomycosis clusters than is currently known. Therefore, pairing genomic data with clinical information and geographic location can be used to monitor blastomycosis infections and determine whether they are clusters, outbreaks, or sporadic occurrences. Our findings demonstrate the utility of genomic analyses for investigating blastomycosis outbreaks, determining genetic diversity of B. dermatitidis and B. gilchristii, and identifying common sources of environmental exposures among cases.
Dr. Bagal is a bioinformatician with the Mycotic Diseases Branch, Division of Foodborne, Waterborne, and Environmental Diseases, National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA. Her research interests are genomics and evolutionary biology, metagenomics, and data science.
Acknowledgments
We thank the Office of Advanced Molecular Detection, National Center for Emerging and Zoonotic Infectious Diseases, CDC, for supporting fungal disease molecular epidemiology; the MDH graduate students who conducted patient interviews; Mitsuru Toda for reviewing and providing feedback, and Suzanne Gibbons-Burgener for providing feedback.
MDH fungal disease epidemiology is supported by the Epidemiology and Laboratory Capacity for Infectious Diseases cooperative agreement with CDC.
References
- Furcolow ML, Busey JF, Menges RW, Chick EW. Prevalence and incidence studies of human and canine blastomycosis. II. Yearly incidence studies in three selected states, 1960-1967. Am J Epidemiol. 1970;92:121–31. DOIPubMedGoogle Scholar
- McTaggart LR, Brown EM, Richardson SE. Phylogeographic analysis of Blastomyces dermatitidis and Blastomyces gilchristii reveals an association with North American freshwater drainage basins. PLoS One. 2016;11:
e0159396 . DOIPubMedGoogle Scholar - Schwartz IS, Wiederhold NP, Hanson KE, Patterson TF, Sigler L. Blastomyces helicus, a new dimorphic fungus causing fatal pulmonary and systemic disease in humans and animals in western Canada and the United States. Clin Infect Dis. 2019;68:188–95. DOIPubMedGoogle Scholar
- Klein BS, Vergeront JM, DiSalvo AF, Kaufman L, Davis JP. Two outbreaks of blastomycosis along rivers in Wisconsin. Isolation of Blastomyces dermatitidis from riverbank soil and evidence of its transmission along waterways. Am Rev Respir Dis. 1987;136:1333–8. DOIPubMedGoogle Scholar
- Reed KD, Meece JK, Archer JR, Peterson AT. Ecologic niche modeling of Blastomyces dermatitidis in Wisconsin. PLoS One. 2008;3:
e2034 . DOIPubMedGoogle Scholar - Thompson K, Sterkel AK, Brooks EG. Blastomycosis in Wisconsin: Beyond the Outbreaks. Acad Forensic Pathol. 2017;7:119–29. DOIPubMedGoogle Scholar
- Chow NA, Gade L, Tsay SV, Forsberg K, Greenko JA, Southwick KL, et al.; US Candida auris Investigation Team. Multiple introductions and subsequent transmission of multidrug-resistant Candida auris in the USA: a molecular epidemiological survey. Lancet Infect Dis. 2018;18:1377–84. DOIPubMedGoogle Scholar
- Oltean HN, Etienne KA, Roe CC, Gade L, McCotter OZ, Engelthaler DM, et al. Utility of whole-genome sequencing to ascertain locally acquired cases of coccidioidomycosis, Washington, USA. Emerg Infect Dis. 2019;25:501–6. DOIPubMedGoogle Scholar
- Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:
e9490 . DOIPubMedGoogle Scholar
Figure
Table
Cite This ArticleOriginal Publication Date: August 10, 2022
Table of Contents – Volume 28, Number 9—September 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Nancy A. Chow, Centers for Disease Control and Prevention, 1600 Clifton Rd NE, Mailstop H17-2, Atlanta, GA 30329-4027, USA
Top