Volume 4, Number 4—December 1998
Synopsis
Cell-to-Cell Signaling and Pseudomonas aeruginosa Infections
Abstract
Pseudomonas aeruginosa is a bacterium responsible for severe nosocomial infections, life-threatening infections in immunocompromised persons, and chronic infections in cystic fibrosis patients. The bacterium's virulence depends on a large number of cell-associated and extracellular factors. Cell-to-cell signaling systems control the expression and allow a coordinated, cell-density–dependent production of many extracellular virulence factors. We discuss the possible role of cell-to-cell signaling in the pathogenesis of P. aeruginosa infections and present a rationale for targeting cell-to-cell signaling systems in the development of new therapeutic approaches.
Figure 1
![Thumbnail of Virulence factors of Pseudomonas aeruginosa. P. aeruginosa has both cell-associated (flagellum, pilus, nonpilus adhesins, alginate/biofilm, lipopolysaccharide [LPS]) and extracellular virulence factors (proteases, hemolysins, exotoxin A, exoenzyme S, pyocyanin).](/eid/images/98-0405-F1-tn.jpg)
Figure 1. Virulence factors of Pseudomonas aeruginosa. P. aeruginosa has both cell-associated (flagellum, pilus, nonpilus adhesins, alginate/biofilm, lipopolysaccharide [LPS]) and extracellular virulence factors (proteases, hemolysins, exotoxin A, exoenzyme S, pyocyanin).
Pseudomonas aeruginosa, an increasingly prevalent opportunistic human pathogen, is the most common gram-negative bacterium found in nosocomial infections. P. aeruginosa is responsible for 16% of nosocomial pneumonia cases (1), 12% of hospital-acquired urinary tract infections (2), 8% of surgical wound infections (3), and 10% of bloodstream infections (4). Immunocompromised patients, such as neutropenic cancer and bone marrow transplant patients, are particularly susceptible to opportunistic infections. In this group of patients, P. aeruginosa is responsible for pneumonia and septicemia with attributable deaths reaching 30% (5,6). P. aeruginosa is also one of the most common and lethal pathogens responsible for ventilator-associated pneumonia in intubated patients (7), with directly attributable death rates reaching 38% (8). In burn patients, P. aeruginosa bacteremia has declined as a result of better wound treatment and dietary changes (removal of raw vegetables, which can be contaminated with P. aeruginosa, from the diet) (3). However, P. aeruginosa outbreaks in burn units are still associated with high (60%) death rates (9). In the expanding AIDS population, P. aeruginosa bacteremia is associated with 50% of deaths (10). Cystic fibrosis (CF) patients are characteristically susceptible to chronic infection by P. aeruginosa, which is responsible for high rates of illness and death in this population (11). The capacity of P. aeruginosa to produce such diverse, often overwhelming infections is due to an arsenal of virulence factors (Figure 1). Many extracellular virulence factors secreted by P. aeruginosa have been shown to be controlled by a complex regulatory circuit involving cell-to-cell signaling systems that allow the bacteria to produce these factors in a coordinated, cell-density–dependent manner (12). In this article we describe major virulence factors of P. aeruginosa and the possible involvement of cell-to-cell signaling in the pathogenesis of acute P. aeruginosa infection. We also summarize data suggesting that these regulatory systems could be exploited for the design of therapeutic interventions.
Figure 2
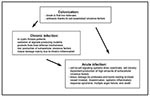
Figure 2. Model of the different phases of Pseudomonas aeruginosa infection. After an initial colonization phase, mostly dependent on cell-associated virulence factors, the infectious process evolves either to a chronic infection characterized by...
P. aeruginosa (family Pseudomonadaceae), an aerobic, motile, gram-negative rod able to grow and survive in almost any environment, lives primarily in water, soil, and vegetation. However, despite abundant opportunities for spread, P. aeruginosa rarely causes community-acquired infections in immunocompetent patients. As a result, the pathogen is viewed as opportunistic. The different phases of P. aeruginosa infection are shown in Figure 2.
Colonization: The Predominant Role of Cell-Associated Virulence Factors
To initiate infection, P. aeruginosa usually requires a substantial break in first-line defenses. Such a break can result from breach or bypass of normal cutaneous or mucosal barriers (e.g., trauma, surgery, serious burns, or indwelling devices), disruption of the protective balance of normal mucosal flora by broad-spectrum antibiotics, or alteration of the immunologic defense mechanisms (e.g., in chemotherapy-induced neutropenia, mucosal clearance defects from cystic fibrosis, AIDS, and diabetes mellitus).
The first step in P. aeruginosa infections is colonization of altered epithelium. The pathogen colonizes the oropharynx of up to 6% and is recovered from the feces of 3% to 24% of healthy persons (2). In contrast, up to 50% of hospitalized patients are at high risk for P. aeruginosa colonization (2). Adherence of P. aeruginosa to epithelium is probably mediated by type 4 pili similar to those of Neisseria gonorrhoeae (13). Several other nonpilus adhesins responsible for the binding to mucin have been described, but their role in the infection process remains unclear (14). Flagella, which are primarily responsible for motility, may also act as adhesins to epithelial cells (15).
From Colonization to Acute Infection: The Role of Extracellular Virulence Factors
P. aeruginosa produces several extracellular products that after colonization can cause extensive tissue damage, bloodstream invasion, and dissemination (Figure 1). In vivo studies have shown that mutants defective in the production of exotoxin A, exoenzyme S, elastase, or alkaline protease are essential for maximum virulence of P. aeruginosa; however, the relative contribution of a given factor may vary with the type of infection (16). Many of these factors are controlled by regulatory systems involving cell-to-cell signaling. We will summarize the known biologic effects of the most-studied extracellular virulence factors associated with acute P. aeruginosa infection.
Exotoxin A is produced by most P. aeruginosa strains that cause clinical infections. Like diphtheria toxin, P. aeruginosa exotoxin A catalyzes ADP-ribosylation and inactivation of elongation factor 2, leading to inhibition of protein biosynthesis and cell death (17). Exotoxin A is responsible for local tissue damage, bacterial invasion (18), and (possibly) immunosuppression (19). Purified exotoxin A is highly lethal for mice which supports its role as a major systemic virulence factor of P. aeruginosa (18).
Exoenzyme S is also an ADP-ribosyl transferase, but unlike exotoxin A, it preferentially ribosylates GTP-binding proteins such as Ras (20). This exoproduct is responsible for direct tissue destruction in lung infection (21) and may be important for bacterial dissemination (22).
Two hemolysins, phospholipase C and rhamnolipid, produced by P. aeruginosa, may act synergistically to break down lipids and lecithin. Both may contribute to tissue invasion by their cytotoxic effects. Rhamnolipid, a rhamnose-containing glycolipid biosurfactant, has a detergentlike structure and is believed to solubilize the phospholipids of lung surfactant, making them more accessible to cleavage by phospholipase C (23). The resulting loss of lung surfactant may be responsible for the atelectasis associated with chronic and acute P. aeruginosa lung infection (24). Rhamnolipid also inhibits the mucociliary transport and ciliary function of human respiratory epithelium (25). However, the relative role of rhamnolipid in acute or chronic infection is not known.
Proteases are assumed to play a major role during acute P. aeruginosa infection. P. aeruginosa produces several proteases including LasB elastase, LasA elastase, and alkaline protease (12). The role of alkaline protease in tissue invasion and systemic infections is unclear; however, its role in corneal infections may be substantial (26,27). The ability of P. aeruginosa to destroy the protein elastin is a major virulence determinant during acute infection. Elastin is a major part of human lung tissue and is responsible for lung expansion and contraction. Moreover, elastin is an important component of blood vessels, which rely on it for their resilience. The concerted activity of two enzymes, LasB elastase and LasA elastase, is responsible for elastolytic activity (28). Elastolytic activity is believed to destroy elastin-containing human lung tissue and cause the pulmonary hemorrhages of invasive P. aeruginosa infections. LasB elastase is a zinc metalloprotease that acts on a number of proteins including elastin. LasB elastase is highly efficient, with a proteolytic activity approximately 10 times that of P. aeruginosa alkaline protease and an activity toward casein approximately four times that of trypsin (28). The LasA elastase is a serine protease that acts synergistically with LasB elastase to degrade elastin. LasA elastase nicks elastin, rendering it sensitive to degradation by other proteases such as LasB elastase, alkaline protease, and neutrophil elastase (28). Both LasB elastase and LasA elastase have been found in the sputum of CF patients during pulmonary exacerbation (29). However, the role of LasB elastase in tissue destruction during the chronic phase of CF is less clear. It has been postulated that during this phase, antibodies present in high titers neutralize LasB elastase, and elastin damaged by minute amounts of LasA is degraded mostly by neutrophil elastase (28). LasB elastase degrades not only elastin but also fibrin and collagen (30). It can inactivate substances such as human immunoglobulins G and A (31), airway lysozyme (32), complement components (33), and substances involved in protecting the respiratory tract against proteases such as a-1-proteinase inhibitor (34) and bronchial mucus proteinase inhibitor (35). Therefore, LasB elastase not only destroys tissue components but also interferes with host defense mechanisms. Studies in animal models show that mutants defective in LasB elastase production are less virulent than their parent strains (16,36,37), which supports the role of LasB elastase as a virulence factor.
Cell-to-Cell Signaling Systems
P. aeruginosa appears to control the production of many of its extracellular virulence factors by a mechanism that monitors bacterial cell density and allows communication between bacteria by cell-to-cell signaling. Bacteria are able to sense their environment, process information, and react appropriately; however, their ability to sense their own cell density, to communicate with each other, and to behave as a population instead of individual cells has only recently been understood (38,39). This phenomenon, called quorum-sensing or cell-to-cell signaling, is a generic phenomenon described in many gram-negative (40) and gram-positive bacteria (41).
Figure 3
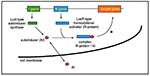
Figure 3. Cell-to-cell signaling systems. Cell-to-cell signaling systems are composed of two genes. The I gene encodes an autoinducer synthase and the R gene encodes a transcriptional activator protein (R-protein). The autoinducer synthase...
The first cell-to-cell signaling system described is the lux system, responsible for the cell-density–dependent control of bioluminescence genes by the marine bacteria Vibrio fischeri (38). Cell-to-cell signaling systems of gram-positive bacteria involve peptide pheromones as signals (41). In contrast, cell-to-cell signaling systems of gram-negative bacteria, with the exception of Ralstonia solanacearum, in which an endogenous fatty acid derivative has been suggested as a cell-to-cell signal (42,43), are composed of a small molecule called an autoinducer, which is synthesized by a LuxI-type autoinducer synthase and a LuxR-type transcriptional activator protein (R-protein) (Figure 3) (38). The various autoinducers described in gram-negative bacteria are homoserine lactone-based molecules that differ between one another in length and substitutions on their acyl side chains. At low cell density, autoinducer is synthesized at basal levels and is thought to diffuse into the surrounding media, where it becomes diluted. With increasing cell density, the intracellular concentration of autoinducer increases until it reaches a threshold concentration. At this critical concentration, the autoinducer has been proposed to bind to a specific R-protein (40). The R-protein itself is not active without the corresponding autoinducer, and it is the R-protein/autoinducer complex that is proposed to bind to specific DNA sequences upstream of target genes enhancing their transcription (44,45). The resulting increase in expression of these genes can reach 1,000-fold. The autoinducer, therefore, allows the bacteria to communicate with each other (cell-to-cell signaling), to sense their own density (quorum-sensing), and together with a transcriptional activator to express specific genes as a population instead of individual cells.
The las Cell-to-Cell Signaling System of P. aeruginosa
Figure 4
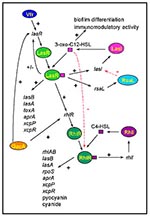
Figure 4. The cell-to-cell signaling circuitry of P. aeruginosa. The las cell-to-cell signaling system controls the rhl cell-to-cell signaling system in a hierarchy cascade. The LasR/3-oxo-C12-HSL complex activates the transcription of rhlR, and...
The first cell-to-cell signaling system described in P. aeruginosa was shown to regulate expression of LasB elastase and was therefore named the las system (46). The las cell-to-cell signaling system is composed of lasI, the autoinducer synthase gene responsible for the synthesis of 3-oxo-C12-HSL (N-[3-oxododecanoyl]-L-homoserine lactone, previously named PAI-1 or OdDHL), and the lasR gene that codes for a transcriptional activator protein (Figure 4) (47,48). The las cell-to-cell signaling system regulates lasB expression and is required for optimal production of other extracellular virulence factors such as LasA protease and exotoxin A (49). LasI is the most sensitive gene to activation by LasR/3-oxo-C12-HSL (50). The preference for the lasI promoter allows an initial rapid rise in autoinducer synthesis, which increases the amount of 3-oxo-C12-HSL available to bind to LasR. This autoinduction hierarchy is responsible for a dramatic increase of expression of virulence genes (such as lasB) once a critical cell density has been reached. Recently, the las system has also been shown to activate the xcpP and xcpR genes that encode proteins of the P. aeruginosa secretory pathway (51). 3-oxo-C12-HSL alone has been suggested to contribute to the virulence of P. aeruginosa because it has some immunomodulatory activity (52). The las cell-to-cell signaling system is positively controlled by GacA (53), as well as by Vfr, which is required for the transcription of lasR (54). An inhibitor, RsaL, that represses the transcription of lasI, has also been described (de Kievit et al., unpub. data). The multiple regulatory levels of the las cell-to-cell signaling system and the various genes under its control highlight the importance of this system for P. aeruginosa.
The rhl Cell-to-Cell Signaling System of P. aeruginosa
P. aeruginosa has a second cell-to-cell signaling system, named the rhl system because of its ability to control the production of rhamnolipid. This system is composed of rhlI, the C4-HSL (N-butyrylhomoserine lactone, previously named PAI-2 or BHL) autoinducer synthase gene, and the rhlR gene encoding a transcriptional activator protein (Figure 4) (55-59). This system regulates the expression of the rhlAB operon that encodes a rhamnosyltransferase required for rhamnolipid production (60). The rhl system is also necessary for optimal production of LasB elastase, LasA protease, pyocyanin, cyanide, and alkaline protease (53,57,61,62). Therefore, like the las cell-to-cell signaling system, the rhl system, sometimes referred to as vsm (virulence secondary metabolites), regulates the expression of various extracellular virulence factors of P. aeruginosa. Interestingly, the rhl system also regulates the expression of rpoS, which encodes a stationary sigma factor (dS) involved in the regulation of various stress-response genes (63,64).
The Cell-to-Cell Signaling Hierarchy in P. aeruginosa
Recent data have shown that the las and rhl cell-to-cell signaling systems of P. aeruginosa interact. Both systems are highly specific in that their respective autoinducers are unable to activate the transcriptional activator protein of the other system (i.e., 3-oxo-C12-HSL cannot activate RhlR, and C4-HSL cannot activate LasR) (57,62). It has also been shown that the R-protein/autoinducer complexes prefer certain promoters that they will activate; LasR/3-oxo-C12-HSL preferentially activates lasB over rhlA, and RhlR/C4-HSL preferentially activates rhlA over lasB (57,62). However, neither system is completely independent of the other. The LasR/3-oxo-C12-HSL complex activates the expression of rhlR placing the las system in a cell-to-cell signaling hierarchy above the rhl system (Figure 4) (63,65). Moreover, 3-oxo-C12-HSL can bind to RhlR, blocking the binding of C4-HSL to its transcriptional activator rhlR (65). The las system therefore controls the rhl system at both a transcriptional and posttranslational level. Another yet unidentified regulatory mechanism directly influencing the expression of rhlRI has been suggested (63). Rhl system regulation of such important genes as rpoS could explain why multiple levels of controls are required for its tight regulation.
Cell-to-Cell Signaling: A Powerful System to Overcome Host Defenses during Acute Infection
Cell-to-cell signaling systems might enable P. aeruginosa to overcome host defense mechanisms. Isolated production of extracellular virulence factors by a small number of bacteria would probably lead to an efficient host response neutralizing these compounds. However, the coordinated expression of virulence genes by an entire bacterial population once a certain density has been reached might allow P. aeruginosa to secrete extracellular factors only when they can be produced at high enough levels to overcome host defenses. These factors could alter the precarious balance between host defenses and production of bacterial toxins, leading to invasion of blood vessels, dissemination, systemic inflammatory-response syndrome, and finally death. Even appropriate antibiotic therapies are often unable to stop this course; therefore, the process must be blocked early, before virulence gene expression can be coordinated.
Biofilms and Cell-to-Cell Signaling
The most striking feature of persistent P. aeruginosa infections in CF patients is the selection of mucoid mutants producing the exopolysaccharide alginate (a polymer of mannuronic and guluronic acid) (11). These mutant bacteria grow inside a biofilm (defined as microcolonies surrounded by an exopolysaccharide) as a survival strategy because the surrounding matrix protects bacteria from phagocytes and complement activity (11). P. aeruginosa growing in an alginate "slime matrix" are resistant to antibiotics (e.g., aminoglycosides, ß-lactam antibiotics, fluoroquinolones) and disinfectants (11). The exact nature of the increased resistance is unclear but has been attributed to slow growth, penetration barriers, ß-lactamase production, and other factors. P. aeruginosa also produces other less well-defined biofilms essential in the colonization of indwelling devices such as catheters. Recently, the las cell-to-cell signaling system has been shown to be involved in the differentiation of P. aeruginosa biofilms (66). A mutant defective in the production of 3-oxo-C12-HSL formed an abnormal biofilm that, in contrast to the wild type biofilm, was sensitive to low concentrations of the detergent sodium dodecyl sulfate (SDS). Furthermore, the addition of 3-oxo-C12-HSL to the culture media restored production of a differentiated, SDS-resistant biofilm by the mutant (66). Whether the formation of an undifferentiated biofilm renders this mutant more sensitive to antibiotics is still unknown. Also unclear is whether 3-oxo-C12-HSL is required for the differentiation of alginate biofilms. The link between 3-oxo-C12-HSL and biofilm differentiation highlights the broad range of systems controlled by cell-to-cell signaling in P. aeruginosa.
Interference with Cell-to-Cell Signaling: A Potential Therapeutic Approach against P. aeruginosa
The increasing emergence of bacterial strains resistant to antibacterial drugs is a major challenge. P. aeruginosa is one of the most problematic human pathogens, showing intrinsic resistance to many structurally unrelated antibiotics. Resistance mechanisms include low outer membrane permeability or multidrug efflux pumps (tetracycline, imipenem, fluoroquinolones, aminoglycosides) or production of antibiotic modifying enzymes (aminoglycosides, ß-lactams) (67,68). Previous exposure to antibiotics often leads to multidrug-resistant P. aeruginosa strains. Moreover, the eradication of colonization with P. aeruginosa is almost impossible in CF patients because of the emergence of multidrug-resistant strains and the protective effects of alginate (69). Therefore, major efforts have been made to find new therapeutic interventions against P. aeruginosa. A tempting approach is to reduce the production of extracellular virulence factors. In this regard, the interference with cell-to-cell signaling systems is very attractive. Experimental evidence for effective interference with cell-to-cell signaling systems has been recently provided by the use of autoinducer analogs (70-72) or furanone compounds (73). The LasR/3-oxo-C12-HSL complex, which plays a central role in controlling virulence gene expression in P. aeruginosa, is essential not only for the production of elastase and other proteases required for tissue invasion but also for the control of the rhl cell-to-cell signaling system in a hierarchical cascade and for the differentiation of biofilm. Mutants with a nonfunctional las cell-to-cell signaling system are unable to produce several virulence factors (including elastase) and have significantly reduced virulence in a neonatal mouse pneumonia model (37). Therefore, the LasR/3-oxo-C12-HSL complex has become a potential target for new therapeutic interventions. Analogs of 3-oxo-C12-HSL (74) are under investigation for their ability to act as 3-oxo-C12-HSL antagonists, which block activation of LasR and thereby interfere with virulence gene expression. The fact that 3-oxo-C12-HSL can block the bioactivity of C4-HSL (65) suggests that it might be possible to interfere efficiently with cell-to-cell signaling in P. aeruginosa. As elements essential for the synthesis of autoinducers have recently been described, drugs inhibiting the biosynthesis process could also be designed (75-80). These approaches could reduce the production of virulence factors, and by interfering with biofilm differentiation they could also render colonizing P. aeruginosa more susceptible to antibiotics and biocides. Combination therapies, including antibiotics and drugs reducing virulence factor production, might have an important role in the treatment of CF and intensive-care patients.
As with any therapeutic approach against P. aeruginosa, the possible emergence of mutations that would prevent the desired effect is a concern. We have recently described spontaneous mutants that were restored in the production of extracellular virulence factors (elastase and rhamnolipid) in the absence of a functional las cell-to-cell signaling system (81). We were unable to isolate mutants restored in virulence factor production when both the las and the rhl cell-to-cell signaling systems were deficient. These results suggest that therapeutic interventions directed only against the las cell-to-cell signaling system are likely to fail because of the emergence of resistant strains able to produce virulence factors despite an inactive las system (81). However, if both the las and the rhl cell-to-cell signaling systems are blocked, P. aeruginosa might be unable to restore the production of cell-to-cell signaling dependent virulence factors; this approach may efficiently reduce virulence factor production and high death rates associated with P. aeruginosa.
Other gram-negative human pathogens including Serratia marcescens, S. liquefaciens, Aeromonas hydrophila, Vibrio cholerae, V. parahaemoliticus, Yersinia enterocolitica, Enterobacter agglomerans, Hafnia alvei, and Rahnella aquatis have been shown to produce homoserine lactone-based autoinducer molecules. These organisms are thought to contain cell-to-cell signaling systems analogous to the las and rhl systems of P. aeruginosa (39,40). Burkholderia cepacia (formerly known as Pseudomonas cepacia) is an opportunistic pathogen responsible for fatal pulmonary infections in CF patients (11). Virulence factor production of this pathogen can be enhanced by P. aeruginosa exoproducts, and this interspecies communication may be due to homoserine-lactone signals (82). Whether B. cepacia contains cell-to-cell signaling systems is still unknown. As the number of bacteria known to use cell-to-cell signaling systems increases, therapeutic interference with cell-to-cell signaling might expand to other human pathogens.
The pathogenesis of P. aeruginosa is clearly multifactorial as underlined by the large number of virulence factors and the broad spectrum of diseases the bacterium causes. Many of the extracellular virulence factors required for tissue invasion and dissemination are controlled by cell-to-cell signaling systems involving homoserine lactone-based signal molecules and specific transcriptional activator proteins. These regulatory systems enable P. aeruginosa to produce virulence factors in a coordinated, cell-density–dependent manner that could allow the bacteria to overwhelm the host defense mechanisms. Interference with cell-to-cell signaling dependent virulence factor production is a promising therapeutic approach for reducing illness and death associated with P. aeruginosa colonization and infection. The growing number of human pathogens found to contain cell-to-cell signaling systems highlights the importance of exploring interference with bacterial cell-to-cell signaling for new therapeutic interventions.
Dr. van Delden is consulting physician, Division of Infectious Diseases, and investigator, Swiss National Science Foundation, Department of Genetics and Microbiology, University Hospital, Geneva, Switzerland. His research interests include bacterial pathogenesis and the development of new antibacterial approaches.
Dr. Iglewski is professor and chair, Department of Microbiology and Immunology, University of Rochester, Rochester, New York. Her research focuses on microbial pathogenesis, cell-to-cell signaling, and gene regulation in bacteria.
Acknowledgments
We thank E.C. Pesci, T. deKievit, and J.P. Pearson for helpful discussions.
Supported by NIH grant R01A133713-04 (to B.H.I.), a Wilmot Foundation, and a Swiss Research Foundation grant (to C.V.D.).
References
- Wiblin RT. Nosocomial pneumonia. In: Wenzel RP, editor. Prevention and control of nosocomial infections. 3rd ed. Baltimore: Williams and Wilkins; 1997. p. 807-19.
- Pollack M. Pseudomonas aeruginosa. In: Mandell GL, Benett JE, Dolin R, editors. Principles and practice of infectious diseases. 4th ed. New York: Churchill Livingstone; 1995. p. 1980-2003.
- Kluytmans J. Surgical infections including burns. In: Wenzel RP, editor. Prevention and control of nosocomial infections. 3rd ed. Baltimore: Williams and Wilkins; 1997. p. 841-65.
- Gordon SM, Serkey JM, Keys TF, Ryan T, Fatica CA, Schmitt SK, Secular trends in nosocomial bloodstream infections in a 55-bed cardiothoracic intensive care unit. Ann Thorac Surg. 1998;65:95–100. DOIPubMedGoogle Scholar
- Fergie JE, Shema SJ, Lott L, Crawford R, Patrick CC. Pseudomonas aeruginosa bacteremia in immunocompromised children: analysis of factors associated with a poor outcome. Clin Infect Dis. 1994;18:390–4.PubMedGoogle Scholar
- Bergen GA, Shelhamer JH. Pulmonary infiltrates in the cancer patient. Infect Dis Clin North Am. 1996;10:297–326. DOIPubMedGoogle Scholar
- Dunn M, Wunderink RG. Ventilator-associated pneumonia caused by Pseudomonas infection [review]. Clin Chest Med. 1995;16:95–109.PubMedGoogle Scholar
- Brewer SC, Wunderink RG, Jones CB, Leeper KVJ. Ventilator-associated pneumonia due to Pseudomonas aeruginosa. Chest. 1996;109:1019–29. DOIPubMedGoogle Scholar
- Richard P, Le FR, Chamoux C, Pannier M, Espaze E, Richet H. Pseudomonas aeruginosa outbreak in a burn unit: role of antimicrobials in the emergence of multiply resistant strains. J Infect Dis. 1994;170:377–83.PubMedGoogle Scholar
- Mendelson MH, Gurtman A, Szabo S, Neibart E, Meyers BR, Policar M, Pseudomonas aeruginosa bacteremia in patients with AIDS [review]. Clin Infect Dis. 1994;18:886–95.PubMedGoogle Scholar
- Govan JR, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkolderia cepacia. Microbiol Rev. 1996;60:539–74.PubMedGoogle Scholar
- Passador L, Iglewski BH. Quorum sensing and virulence gene regulation in Pseudomonas aeruginosa. In: Roth JA, editor. Virulence mechanisms of bacterial pathogens. 2nd ed. Washington: American Society for Microbiology; 1995. p. 65-78.
- de Bentzmann S, Roger P, Bajolet-Laudinat O, Fuchey C, Plotkowski MC, Puchell E. Asialo GM1 is a receptor for Pseudomonas aeruginosa adherence to regenerating respiratory epithelial cells. Infect Immun. 1996;64:1582–8.PubMedGoogle Scholar
- Gilboa-Garber N. Towards anti-Pseudomonas aeruginosa adhesion therapy. In: Kahane O, editor. Toward anti-adhesion therapy for microbial diseases. New York: Plenum Press; 1996. p. 39-50.
- Feldman M, Bryan R, Rajan S, Scheffler L, Brunnert S, Tang H, Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect Immun. 1998;66:43–51.PubMedGoogle Scholar
- Nicas TI, Iglewski BH. The contribution of exoproducts to virulence of Pseudomonas aeruginosa. Can J Microbiol. 1985;31:387–92.PubMedGoogle Scholar
- Wick MJ, Hamood AN, Iglewski BH. Analysis of the structure-function relationship of Pseudomonas aeruginosa exotoxin A [review]. Mol Microbiol. 1990;4:527–35. DOIPubMedGoogle Scholar
- Woods DE, Iglewski BH. Toxins of Pseudomonas aeruginosa: new perspectives [review]. Rev Infect Dis. 1983;5(Suppl 4):S715–22.PubMedGoogle Scholar
- Vidal DR, Garrone P, Banchereau J. Immunosuppressive effects of Pseudomonas aeruginosa exotoxin A on human B-lymphocytes. Toxicon. 1993;31:27–34. DOIPubMedGoogle Scholar
- Iglewski BH, Sadoff J, Bjorn MJ, Maxwell ES. Pseudomonas aeruginosa exoenzyme S: an adenosine diphosphate ribosyltransferase distinct from toxin A. Proc Natl Acad Sci U S A. 1978;75:3211–5. DOIPubMedGoogle Scholar
- Nicas TI, Frank DW, Stenzel P, Lile JD, Iglewski BH. Role of exoenzyme S in chronic Pseudomonas aeruginosa lung infections. Eur J Clin Microbiol. 1985;4:175–9. DOIPubMedGoogle Scholar
- Nicas TI, Bradley J, Lochner JE, Iglewski BH. The role of exoenzyme S in infections with Pseudomonas aeruginosa. J Infect Dis. 1985;152:716–21.PubMedGoogle Scholar
- Liu PV. Extracellular toxins of Pseudomonas aeruginosa. J Infect Dis. 1974;130:94–9.
- Liu PV. Toxins of Pseudomonas aeruginosa. In: Doggett RG, editor. Pseudomonas aeruginosa. Clinical manifestations of infection and current therapy. New York: Academic Press; 1979. p. 63-88.
- Read RC, Roberts P, Munro N. Effect of Pseudomonas aeruginosa rhamnolipids on mucociliary transport and ciliary beating. J Appl Physiol. 1992;72:2271–7.PubMedGoogle Scholar
- Kernacki KA, Hobden JA, Hazlett LD, Fridman R, Berk RS. In vivo bacterial protease production during Pseudomonas aeruginosa corneal infection [published erratum appears in Invest Ophthalmol Vis Sci 1995;36:1947]. Invest Ophthalmol Vis Sci. 1995;36:1371–8.PubMedGoogle Scholar
- Howe TR, Iglewski BH. Isolation and characterization of alkaline protease-deficient mutants of Pseudomonas aeruginosa in vitro and in a mouse eye model. Infect Immun. 1984;43:1058–63.PubMedGoogle Scholar
- Galloway DR. Pseudomonas aeruginosa elastase and elastolysis revisited: recent developments. Mol Microbiol. 1991;5:2315–21. DOIPubMedGoogle Scholar
- Jaffar-Bandjee MC, Lazdunski A, Bally M, Carrere J, Chazalette JP, Galabert C. Production of elastase, exotoxin A, and alkaline protease in sputa during pulmonary exacerbation of cystic fibrosis in patients chronically infected by Pseudomonas aeruginosa. J Clin Microbiol. 1995;33:924–9.PubMedGoogle Scholar
- Heck LW, Morihara K, McRae WB, Miller EJ. Specific cleavage of human type III and IV collagens by Pseudomonas aeruginosa elastase. Infect Immun. 1986;51:115–8.PubMedGoogle Scholar
- Heck LW, Alarcon PG, Kulhavy RM, Morihara K, Russell MW, Mestecky JF. Degradation of IgA proteins by Pseudomonas aeruginosa elastase. J Immunol. 1990;6:2253–7.
- Jacquot J, Tournier J, Puchelle E. In vitro evidence that human airway lysozyme is cleaved and inactivated by Pseudomonas aeruginosa elastase and not by human leukocyte elastase. Infect Immun. 1985;47:555–60.PubMedGoogle Scholar
- Hong Y, Ghebrehiwet B. Effect of Pseudomonas aeruginosa elastase and alkaline protease on serum complement and isolated components C1q and C3. Clin Immunol Immunopathol. 1992;62:133–8. DOIPubMedGoogle Scholar
- Morihara K, Tsuzuki H, Oda K. Protease and elastase of Pseudomonas aeruginosa: inactivation of human a 1-proteinase inhibitor. Infect Immun. 1979;24:188–93.PubMedGoogle Scholar
- Johnson DA, Carter-Hamm B, Dralle WM. Inactivation of human bronchial mucous proteinase inhibitor by Pseudomonas aeruginosa elastase. Am Rev Respir Dis. 1982;126:1070–3.PubMedGoogle Scholar
- Tamura Y, Suzuki S, Sawada T. Role of elastase as a virulence factor in experimental Pseudomonas aeruginosa infection in mice. Microb Pathog. 1992;12:237–44. DOIPubMedGoogle Scholar
- Tang HB, DiMango E, Bryan R, Gambello M, Iglewski BH, Goldberg JB, Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect Immun. 1996;64:37–43.PubMedGoogle Scholar
- Fuqua C, Winans SC, Greenberg EP. Census and consensus in bacterial ecosystems: the LuxR-LuxI family of quorum-sensing transcriptional regulators [review]. Annu Rev Microbiol. 1996;50:727–51. DOIPubMedGoogle Scholar
- Gray KM. Intercellular communication and group behaviour in bacteria. Trends Microbiol. 1997;5:184–8. DOIPubMedGoogle Scholar
- Greenberg EP. Quorum sensing in gram-negative bacteria. ASM News. 1997;63:371–7.
- Kleerebezem M, Quadri LE, Kuipers OP, de Vos VM. Quorum sensing by peptide pheromones and two-component signal-transduction systems in gram-positive bacteria [review]. Mol Microbiol. 1997;24:895–904. DOIPubMedGoogle Scholar
- Clough SJ, Lee KE, Schell MA, Denny TP. A two-component system in Ralstonia (Pseudomonas) solanacearum modulates production of PhcA-regulated virulence factors in response to 3-hydroxypalmitic acid methyl ester. J Bacteriol. 1997;179:3639–48.PubMedGoogle Scholar
- Flavier AB, Clough SJ, Schell MA, Denny TP. Identification of 3-hydroxypalmitic acid methyl ester as a novel autoregulator controlling virulence in Ralstonia solanacearum. Mol Microbiol. 1997;26:251–9. DOIPubMedGoogle Scholar
- Stevens AM, Dolan KM, Greenberg EP. Synergistic binding of the Vibrio fischeri LuxR transcriptional activator domain and RNA polymerase to the lux promoter region [published erratum appears in Proc Natl Acad Sci U S A 1995;92:3631]. Proc Natl Acad Sci U S A. 1994;91:12619–23. DOIPubMedGoogle Scholar
- Stevens AM, Greenberg EP. Quorum sensing in Vibrio fischeri: essential elements for activation of the luminescence genes. J Bacteriol. 1997;179:557–62.PubMedGoogle Scholar
- Passador L, Cook JM, Gambello MJ, Rust L, Iglewski BH. Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science. 1993;260:1127–30. DOIPubMedGoogle Scholar
- Pearson JP, Gray KM, Passador L, Tucker KD, Eberhard A, Iglewski BH, Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc Natl Acad Sci U S A. 1994;91:197–201. DOIPubMedGoogle Scholar
- Gambello MJ, Iglewski BH. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J Bacteriol. 1991;173:3000–9.PubMedGoogle Scholar
- Gambello MJ, Kaye S, Iglewski BH. LasR of Pseudomonas aeruginosa is a transcriptional activator of the alkaline protease gene (apr) and an enhancer of exotoxin A expression. Infect Immun. 1993;61:1180–4.PubMedGoogle Scholar
- Seed PC, Passador L, Iglewski BH. Activation of the Pseudomonas aeruginosa lasI gene by LasR and the Pseudomonas autoinducer PAI: an autoinduction regulatory hierarchy. J Bacteriol. 1995;177:654–9.PubMedGoogle Scholar
- Chapon V, Akrim M, Latifi A, Williams P, Lazdunski A, Bally M. Regulation of the xcp secretion pathway by multiple quorum-sensing modulons in Pseudomonas aeruginosa. Mol Microbiol. 1997;24:1169–78. DOIPubMedGoogle Scholar
- Telford G, Wheeler D, Williams P, Tomkins PT, Appleby O, Sewell H, The Pseudomonas aeruginosa quorum-sensing signal molecule N-(3-oxododecanoyl)-L-homoserine lactone has immunomodulatory activity. Infect Immun. 1998;66:36–42.PubMedGoogle Scholar
- Reimmann C, Beyeler M, Latifi A, Winteler H, Foglino M, Lazdunski A, The global activator GacA of Pseudomonas aeruginosa PAO positively controls the production of the autoinducer N-butyryl-homoserine lactone and the formation of the virulence factors pyocyanin, cyanide, and lipase. Mol Microbiol. 1997;24:309–19. DOIPubMedGoogle Scholar
- Albus AM, Pesci EC, Runyen-Janecky LJ, West SE, Iglewski BH. Vfr controls quorum sensing in Pseudomonas aeruginosa. J Bacteriol. 1997;179:3928–35.PubMedGoogle Scholar
- Pearson JP, Passador L, Iglewski BH, Greenberg EP. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 1995;92:1490–4. DOIPubMedGoogle Scholar
- Ochsner UA, Koch AK, Fiechter A, Reiser J. Isolation and characterization of a regulatory gene affecting rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. J Bacteriol. 1994;176:2044–54.PubMedGoogle Scholar
- Latifi A, Winson MK, Foglino M, Bycroft BW, Stewart GS, Lazdunski A, . Multiple homologues of LuxR and LuxI control expression of virulence determinants and secondary metabolites through quorum sensing in Pseudomonas aeruginosa PAO1. Mol Microbiol. 1995;17:333–43. DOIPubMedGoogle Scholar
- Winson MK, Camara M, Latifi A, Foglino M, Chhabra SR, Daykin M, Multiple N-acyl-L-homoserine lactone signal molecules regulate production of virulence determinants and secondary metabolites in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 1995;92:9427–31. DOIPubMedGoogle Scholar
- Ochsner UA, Reiser J. Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 1995;92:6424–8. DOIPubMedGoogle Scholar
- Ochsner UA, Fiechter A, Reiser J. Isolation, characterization, and expression in Escherichia coli of the Pseudomonas aeruginosa rhlAB genes encoding a rhamnosyltransferase involved in rhamnolipid biosurfactant synthesis. J Biol Chem. 1994;269:19787–95.PubMedGoogle Scholar
- Brint JM, Ohman DE. Synthesis of multiple exoproducts in Pseudomonas aeruginosa is under the control of RhlR-RhlI, another set of regulators in strain PAO1 with homology to the autoinducer-responsive LuxR-LuxI family. J Bacteriol. 1995;177:7155–63.PubMedGoogle Scholar
- Pearson JP, Pesci EC, Iglewski BH. Role of Pseudomonas aeruginosa las and rhl quorum-sensing systems in the control of elastase and rhamnolipid biosynthesis genes. J Bacteriol. 1997;179:5756–67.PubMedGoogle Scholar
- Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol Microbiol. 1996;21:1137–46. DOIPubMedGoogle Scholar
- Loewen PC, Hengge-Aronis R. The role of the sigma factor sigma S (KatF) in bacterial global regulation [review]. Annu Rev Microbiol. 1994;48:53–80. DOIPubMedGoogle Scholar
- Pesci EC, Pearson JP, Seed PC, Iglewski BH. Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol. 1997;179:3127–32.PubMedGoogle Scholar
- Davies DG, Parsek MR, Pearson JP, Iglewski BH, Costerton JW, Greenberg EP. The involvement of cell-to-cell signals in the development of bacterial biofilm. Science. 1998;280:295–8. DOIPubMedGoogle Scholar
- Nakae T. Role of membrane permeability in determining antibiotic resistance in Pseudomonas aeruginosa. Microbiol Immunol. 1995;39:221–9.PubMedGoogle Scholar
- Nikaido H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996;178:5853–9.PubMedGoogle Scholar
- Hoiby N, Giwercman B, Jensen ET, Pedersen SS, Koch C, Kharazmi A. Mechanism of action of antibiotics in chronic pulmonary Pseudomonas infection [review]. Adv Pharmacol. 1994;30:53–84. DOIPubMedGoogle Scholar
- Milton DL, Hardman A, Camara M, Chhabra SR, Bycroft BW, Stewart GS, Quorum sensing in Vibrio anguillarum: characterization of the vanI/vanR locus and identification of the autoinducer N-(3- oxodecanoyl)-L-homoserine lactone. J Bacteriol. 1997;179:3004–12.PubMedGoogle Scholar
- McClean KH, Winson MK, Fish L, Taylor A, Chhabra SR, Camara M, Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology. 1997;143:3703–11. DOIPubMedGoogle Scholar
- Swift S, Karlyshev AV, Fish L, Durant EL, Winson MK, Chhabra SR, Quorum sensing in Aeromonas hydrophila and Aeromonas salmonicida: identification of the LuxRI homologs AhyRI and AsaRI and their cognate N-acylhomoserine lactone signal molecules. J Bacteriol. 1997;179:5271–81.PubMedGoogle Scholar
- Givskov M, de Nys R, Manefield M, Gram L, Maximilien R, Eberl L, Eukaryotic interference with homoserine lactone-mediated prokaryotic signalling. J Bacteriol. 1996;178:6618–22.PubMedGoogle Scholar
- Passador L, Tucker KD, Guertin KR, Journet MP, Kende AS, Iglewski BH. Functional analysis of the Pseudomonas aeruginosa autoinducer PAI. J Bacteriol. 1996;178:5995–6000.PubMedGoogle Scholar
- Jiang Y, Camara M, Chhabra SR, Hardie KR, Bycroft BW, Lazdunski A, In vitro biosynthesis of the Pseudomonas aeruginosa quorum-sensing signal molecule N-butanoyl-L-homoserine lactone. Mol Microbiol. 1998;28:193–203. DOIPubMedGoogle Scholar
- Val DL, Cronan JEJ. In vivo evidence that S-adenosylmethionine and fatty acid synthesis intermediates are the substrates for the LuxI family of autoinducers. J Bacteriol. 1998;180:2644–51.PubMedGoogle Scholar
- Schaefer AL, Val DL, Hanzelka BL, Cronan JEJ, Greenberg EP. Generation of cell-to-cell signals in quorum sensing: acyl homoserine lactone synthase activity of a purified Vibrio fischeri LuxI protein. Proc Natl Acad Sci U S A. 1996;93:9505–9. DOIPubMedGoogle Scholar
- More MI, Finger LD, Stryker JL, Fuqua C, Eberhard A, Winans SC. Enzymatic synthesis of a quorum-sensing autoinducer through use of defined substrates. Science. 1996;272:1655–8. DOIPubMedGoogle Scholar
- Hanzelka BL, Stevens AM, Parsek MR, Crone TJ, Greenberg EP. Mutational analysis of the Vibrio fischeri LuxI polypeptide: critical regions of an autoinducer synthase. J Bacteriol. 1997;179:4882–7.PubMedGoogle Scholar
- Parsek MR, Schaefer AL, Greenberg EP. Analysis of random and site-directed mutations in rhlI, a Pseudomonas aeruginosa gene encoding an acylhomoserine lactone synthase. Mol Microbiol. 1997;26:301–10. DOIPubMedGoogle Scholar
- Van Delden C, Pesci EC, Pearson JP, Iglewski BH. Starvation selection restores elastase and rhamnolipid production in a Pseudomonas aeruginosa quorum-sensing mutant. Infect Immun. 1998;66:4478–502.
- McKenney D, Brown KE, Allison DG. Influence of Pseudomonas aeruginosa exoproducts on virulence factor production in Burkholderia cepacia: evidence of interspecies communication. J Bacteriol. 1995;177:6989–92.PubMedGoogle Scholar
Figures
Cite This ArticleTable of Contents – Volume 4, Number 4—December 1998
| EID Search Options |
|---|
|
|
|
|
|
|