Volume 15, Number 10—October 2009
Dispatch
Fine-scale Identification of the Most Likely Source of a Human Plague Infection
Abstract
We describe an analytic approach to provide fine-scale discrimination among multiple infection source hypotheses. This approach uses mutation-rate data for rapidly evolving multiple locus variable-number tandem repeat loci in probabilistic models to identify the most likely source. We illustrate the utility of this approach using data from a North American human plague investigation.
Linking human disease events to likely sources of infection has been advanced by molecular epidemiology. However, isolates from several potential infection sources often are similar, and none may exactly match the clinical isolate genotype, especially if the methods used provide high discrimination (1). Conclusions from partial-match genotypes are problematic but may provide the only data for weighing the relative importance of similar source genotypes. Even perfect-match genotypes do not preclude partial-match sources as likely infection sources (2). We present a probabilistic approach based on mutation rates that can be used to identify the most likely source of infection. Our example is human plague, but the approach could be applied to other diseases for which data on marker mutation rates are available (3).
Plague is caused by the bacterium Yersinia pestis. Because Y. pestis is an obligate pathogen that continuously cycles between rodents and fleas, mutations are generated regularly and can be observed among even closely related isolates (1). Human contact with infected fleas or rodents can result in human plague (4). Plague is rare in the United States, with <20 cases in 2006 (5) but is of concern because of the potential use of Y. pestis as a biological weapon (6). Thus, the ability to link a human plague isolate to a likely source has implications for investigating both natural disease and bioterrorism events.
Multiple locus variable-number tandem repeat (VNTR) analysis (MLVA) is useful for molecular epidemiologic studies of Y. pestis because of its discrimination power (1,7,8). We previously used MLVA to genotype the human isolate described below and queried the resulting genotype against a database containing genotypes from hundreds of Y. pestis isolates (9). This statistical approach identified isolates that most closely matched the human isolate and confirmed its most likely coarse geographic origin (northern New Mexico). However, this set of near matches from the database query included isolates representing several different potential local infection sources, leaving the most likely fine-scale source unclear. The human and environmental isolates were indistinguishable with pulsed-field gel electrophoresis (PFGE); thus, the most likely fine-scale source could not be identified (10).
In November 2002, while visiting New York, New York, USA, 2 persons from Santa Fe County, New Mexico, USA, became ill with fever and unilateral inguinal adenopathy; clinicians subsequently identified the illness as bubonic plague. Investigation by the New Mexico Department of Health and the Centers for Disease Control and Prevention indicated the patients were infected in New Mexico because Y. pestis–positive fleas were collected near the patients’ home (10). However, because plague is endemic to the region, and flea samples from which isolates were obtained were collected at the home and along a local trail on which the patients hiked, either location could be the source. To identify the most likely fine-scale source of their infections, we examined specific mutations separating the human isolate from closely related environmental isolates.
Figure 1
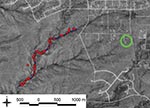
Figure 1. Distribution of rodent trapping stations along a hiking trail in Santa Fe County, New Mexico, USA. Each red circle indicates a single trapping site that had 3 traps. Trap stations (not...
We examined 5 Y. pestis isolates (Table 1) to develop a fine-scale spatial analysis of the infection. The reference isolate was obtained from 1 patient, 3 isolates were obtained from fleas collected in the patients’ yard (9) (2 were collected before their illness as part of a long-term investigation), and 1 isolate was obtained from the trail flea samples a short time later as part of the same long-term study (Figure 1). Other isolates were collected and examined but were excluded from this fine-scale analysis because they were more distinct from the human isolate, differing at >4 VNTR loci. DNA extracts were prepared from each isolate (11,12) and analyzed using a 43-loci MLVA system as previously described (1,8).
Figure 2
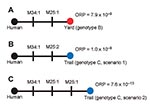
Figure 2. Alternate infection source hypotheses for the plague cases in the persons who visited New York, New York, USA. Closed circles indicate genotypes; black, red, and blue circles indicate genotypes A, B,...
We observed 3 MLVA genotypes (A–C) among the 5 samples (Table 1, Figure 2). The human isolate was assigned genotype A. Genotype B, observed in 3 isolates obtained from the yard, differed from the reference by single-repeat mutations at 2 VNTR loci (M25 and M34; Figure 2, panel A). Genotype C, observed in 1 isolate from a flea obtained along the trail, also differed from the reference isolate at loci M25 and M34. However, the mutation at M25 was a double-repeat mutation that could be explained 2 ways: as a single 2-repeat mutational event (Figure 2, panel B) or as 2 sequential single-repeat mutations at the same locus (Figure 2, panel C). Although all 43 VNTR loci are useful for identifying the coarse geographic origin of an unknown isolate by using a database approach (9), our analysis examined only polymorphic loci because monomorphic loci provided no additional information. The molecular epidemiologic goal was to identify the environmental isolate most closely related to the human isolate and thus the most likely fine-scale geographic source of the infection.
To this end, we examined the relative probability of each mutation (Table 2) using published mutation rate data (1,8). We used mutation rate estimates for specific mutational events to judge relative probabilities of different scenarios. This approach assumes 1) there is an intrinsic mutation rate at each loci for each event, 2) we have accurately estimated these rates (1,8), and 3) we can use intrinsic rates to judge the relative likelihood of >2 hypotheses. We multiplied individual probabilities of mutations within a scenario to calculate the overall relative probability (ORP) that an environmental isolate was related to the infection source (Table 2; Figure 2). To select the most likely source, we compared the ORP of each scenario with the others in a pairwise fashion (odds ratios, Table 2). In practice, only the most likely source needs to be compared with all other sources.
The patients most likely were infected from a source in their yard. Genotype B was observed in isolates from the yard, and this scenario had the highest ORP (7.9 × 10–9; hypothesis B→A; Table 2). The first scenario for genotype C (C1→A; Table 2) is second most likely (ORP 1.0 × 10–9). The odds ratio shows the most likely scenario (B→A) is just 7.9× more likely than this scenario (C1→A). These 2 near matches illustrate the power of this approach: one is the most likely source, but the other is statistically possible because this odds ratio difference would not be significant at α<0.05 (odds ratio >20). However, the ORP (1.0 × 104) for the second scenario for genotype C (C2→A; Table 2) would be statistically significant, enabling it to be rejected.
When a high-resolution typing approach based on loci with fast mutation rates, such as MLVA, is used, near matches should be the rule rather than the exception. After transmission, the pathogen will continue to propagate in environmental sources and in the patient, leading to additional mutations before investigators obtain isolates. Mutations may also occur during routine laboratory procedures (e.g., culturing) before genotypic comparisons. Thus, perfect matches are rarely observed during phylogenetic analysis. Rather, the common ancestor (i.e., genotype of the source strain at time of infection) of the human isolate and each potential source isolate will need to be hypothesized. MLVA and probabilistic modeling provide a rigorous means to identify the most likely fine-scale environmental source. The same principles can be applied to other subtyping approaches used in investigations, including those with slower evolution patterns such as PFGE. In these cases, matches and near matches also should be judged by their relative evolutionary rates. Applying evolutionary probabilistic modeling to subtyping will generate stronger conclusions by evaluating the relative strengths of alternative hypotheses regardless of the subtyping approach.
Ms Colman is completing her PhD degree in biological sciences at the Center for Microbial Genetics and Genomics, Northern Arizona University. Her primary research interest is the examination of plague from evolutionary, ecological, and public health standpoints.
Acknowledgment
This work was supported by the National Institutes of Health–National Institute of Allergy and Infectious Diseases (grant 1R15AI070183), the Pacific-Southwest Regional Center of Excellence (AI065359), the Department of Homeland Security Science and Technology Directorate (contract no. HSHQDC-08-C-00158), Achievement Rewards for College Scientists Foundation Inc., and the Cowden Endowment at Northern Arizona University.
References
- Girard JM, Wagner DM, Vogler AJ, Keys C, Allender CJ, Drickamer LC, Differential plague-transmission dynamics determine Yersinia pestis population genetic structure on local, regional, and global scales. Proc Natl Acad Sci U S A. 2004;101:8408–13. DOIPubMedGoogle Scholar
- Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol. 1995;33:2233–9.PubMedGoogle Scholar
- Vogler AJ, Keys C, Nemoto Y, Colman RE, Jay Z, Keim P. Effect of repeat copy number on variable-number tandem repeat mutations in Escherichia coli O157:H7. J Bacteriol. 2006;188:4253–63. DOIPubMedGoogle Scholar
- Perry RD, Fetherston JD. Yersinia pestis—etiologic agent of plague. Clin Microbiol Rev. 1997;10:35–66.PubMedGoogle Scholar
- Centers for Disease Control and Prevention. Human plague—four states, 2006. MMWR Morb Mortal Wkly Rep. 2006;55:940–3.PubMedGoogle Scholar
- Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Plague as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA. 2000;283:2281–90. DOIPubMedGoogle Scholar
- Pourcel C, Andre-Mazeaud F, Neubauer H, Ramisse F, Vergnaud G. Tandem repeats analysis for the high resolution phylogenetic analysis of Yersinia pestis. BMC Microbiol. 2004;4:22. DOIPubMedGoogle Scholar
- Vogler AJ, Keys CE, Allender C, Bailey I, Girard J, Pearson T, Mutations, mutation rates, and evolution at the hypervariable VNTR loci of Yersinia pestis. Mutat Res. 2007;616:145–58. DOIPubMedGoogle Scholar
- Lowell JL, Wagner DM, Atshabar B, Antolin MF, Vogler AJ, Keim P, Identifying sources of human exposure to plague. J Clin Microbiol. 2005;43:650–6. DOIPubMedGoogle Scholar
- Centers for Disease Control and Prevention. Imported plague—New York City, 2002. MMWR Morb Mortal Wkly Rep. 2003;52:725–8.PubMedGoogle Scholar
- Allender CJ, Easterday WR, Van Ert MN, Wagner DM, Keim P. High-throughput extraction of arthropod vector and pathogen DNA using bead milling. Biotechniques. 2004;37:730, 732, 734.PubMedGoogle Scholar
- Keim P, Price LB, Klevytska AM, Smith KL, Schupp JM, Okinaka R, Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. J Bacteriol. 2000;182:2928–36. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 15, Number 10—October 2009
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
David M. Wagner, Northern Arizona University, Flagstaff, AZ 86011-5640, USA
Top