Volume 16, Number 2—February 2010
Research
Associations between Mycobacterium tuberculosis Strains and Phenotypes
Abstract
To inform development of tuberculosis (TB) control strategies, we characterized a total of 2,261 Mycobacterium tuberculosis complex isolates by using multiple phenotypic and molecular markers, including polymorphisms in repetitive sequences (spoligotyping and variable-number tandem repeats [VNTRs]) and large sequence and single-nucleotide polymorphisms. The Beijing family was strongly associated with multidrug resistance (p = 0.0001), and VNTR allelic variants showed strong associations with spoligotyping families: >5 copies at exact tandem repeat (ETR) A, >2 at mycobacterial interspersed repetitive unit 24, and >3 at ETR-B associated with the East African–Indian and M. bovis strains. All M. tuberculosis isolates were differentiated into 4 major lineages, and a maximum parsimony tree was constructed suggesting a more complex phylogeny for M. africanum. These findings can be used as a model of pathogen global diversity.
Tuberculosis (TB), caused by bacteria of the Mycobacterium tuberculosis complex (MTBC), remains a global threat to human health, which causes an estimated 2 million deaths annually (1). No horizontal gene transfer has been reported in MTBC, and the genome is more highly conserved than other pathogenic bacteria (2). Nevertheless, genotyping tools have recently identified several polymorphisms in the MTBC genome that have provided insight into its evolution. Three major groups of MTBC genome alterations have been reported: single nucleotide polymorphisms (SNPs), large sequence polymorphisms (LSPs), and polymorphisms within repetitive sequences such as variable number tandem repeats (VNTRs). The first 2 groups mark irreversible genetic events and can be used to construct phylogenies for M. tuberculosis (2–6). An association between geographic region and M. tuberculosis families, defined by specific polymorphisms, has been demonstrated. This geographic structuring producing genetically, and perhaps phenotypically, distinct MTBC populations may contribute to differences in clinical features such as severity of disease or prevalence of extrapulmonary disease (6–8) and should be considered during the development of new drugs and vaccines.
Sreevatsan et al. divided MTBC strains into 3 principal genetic groups (PGG1–PGG3) based on SNPs in codon 463 of katG and codon 95 of gyrA (2). More recently, on the basis of polymorphisms in the oxyR, katG, and rpoB genes, strains have been divided into 5 lineages (I–IV and M. bovis); lineages I, III, and IV represent subgroups within PGG1, and lineage II corresponds to PGG 2 and 3 (7). By combining these markers with LSPs RD239, RD105, RD750, RD711, and RD702, a small 7bp deletion in the pks15/1 gene and other SNPs, Gagneaux and Small were able to confirm these M. tuberculosis lineages and 2 lineages of M. africanum (6). The deletions RD9 and TbD1 are useful phylogenetic markers for other members of MTBC complex and ancestral M. tuberculosis strains (3). The loss and acquisition of repeats or spacers in the direct repeats region (9) does not appear to limit their value in biogeographic and phylogenetic studies (10,11).
Genotypic variation of MTBC strains at various geographic settings and significant associations between certain allelic variants at VNTR loci, MTBC lineages, and spoligotyping families have been reported (7,12–15). However, most studies used single genotyping methods on small populations or convenience samples. Population-based studies have focused primarily on areas of low- to middle-TB incidence, and it is unclear whether the results are universally applicable (16–18). Larger population-based studies on geographically diverse populations are needed to establish the phylogenetic, epidemiologic, and clinical relevance of such associations.
London accounts for nearly half of all TB cases in the United Kingdom (≈3,300 cases in 2006; incidence rate 44.8/100,000). Because 75% of these TB patients were born abroad (19), (Health Protection Agency update; www.hpa.org.uk), and clinical signs of disease develop within 2 to 3 years of arrival, we believe that the multicultural and diverse community in London provides a unique setting for studying the global biodiversity of MTBC. We aimed to establish whether MTBC isolates circulating in the London population are a useful model of global diversity, to determine the phylogenetic relevance of polymorphisms in repetitive regions of the MTBC genome, especially for M. africanum and its position in TB evolution, and to investigate associations between lineage and phenotype.
Study Design and Bacterial Isolates
One isolate from each of the 2,261 MTBC culture-positive patients was included in this prospectively designed population study. These isolates were collected from patients in all 30 London National Health Service hospitals between April 1, 2005, and March 31, 2006. Demographic data, including gender, date of birth, and country of birth were assigned to world regions according to an existing United Nations classification (20).
Identification
Cultures were identified by using standard phenotypic identification tests (21) and molecular methods (Genotype Mycobacterium CM, AS, and MTBC kits; Hain Lifescience GmbH, Nehren, Germany) and the INNO LiPA Rif TB assay (Innogenetics, Ghent, Belgium) performed as recommended by the manufacturer. DNA was extracted from cultures using chloroform extraction as described (22). Isoniazid, rifampin, ethambutol, streptomycin, pyrazinamide, and ciprofloxacin susceptibilities were determined by using the resistance ratio method (21).
Genotyping
All extracts were typed by using automated 15 mycobacterial interspersed repetitive unit–VNTR (MIRU-VNTR) fragment analysis (23–26). Clustered isolates were further genotyped by using an extended panel of 7 hypervariable VNTR loci (27). Data were exported to BioNumerics (Applied Maths, Sint-Martens-Latem, Belgium) for cluster analysis.
Spoligotyping was performed according to the manufacturer’s instructions (Isogen Lifescience, IJsselstein, the Netherlands) (9). Images were digitized and entered into BioNumerics software by using the BNIMA module (Applied Maths). Spoligotypes were assigned to families and subfamilies by using the online tools at http://cgi2.cs.rpi.edu/~bennek/SPOTCLUST.html (10). We have used the established spoligotyping families Beijing, Central Asian (CAS), East African–Indian (EAI), and M. bovis as lineage designations, as well as European American (EuroAm) (13,28) for the M. tuberculosis lineage, which includes the X, T, LAM, S, and Haarlem families.
Other Methods
Detection of TbD1 and RD9 (3,13) was conducted by PCR fragment analysis (3). Reverse hybridization methods were used to analyze the 4 lineage-defining SNPs in 3 genes (oxyRC37T, katGC87A, rpoBT2646G, and rpoBC3243T) reported by Baker et al. (7) for selected isolates (n = 259) (12) and mutations in katG, inhA, and rpoB genes associated with drug resistance (22).
Data were analyzed by using Excel, BioNumerics (Applied Maths), SPSS 12.0 (SPSS Inc, Chicago, IL, USA) software and online interactive statistical tools (www.quantitativeskills.com/sisa). Categorical variables were analyzed by using relative risks (RRs), odds ratios (ORs), and the χ2 test. Discrimination power of genotyping methods was assessed using the Hunter-Gaston index (29).
Diversity within the Study Population
We studied 2,261 isolates, representing 95.7% of all the bacteriologically confirmed TB cases reported in London from April 1, 2005, through March 31, 2006. Using routine phenotypic and genotypic methods, we identified 99.1% (2,241) as MTBC; the remaining 20 were too heavily contaminated for analysis.
Spoligotypes were generated for 98.8% (2,233) of the isolates; 656 types were identified, of which 458 were unique and 198 were shared by groups of 2–221 isolates. Isolates were assigned to families and subfamilies on the basis of their spoligotype by using the online tools at http://cgi2.cs.rpi.edu/~bennek/SPOTCLUST.html. All but 4 spoligotypes were assigned to >1 of 36 groups; 88.4% of isolates were assigned to a single spoligotyping family or subfamily. The remaining 11.6% were assigned to 2 families, albeit with given probabilities of <0.9. All the main spoligofamilies seen globally were represented within this population (Table 1).
Isolates were cultured from a variety of body sites; 57% were of pulmonary origin. Where known, 60% of isolates were cultured from male patients and 40% from female patients; median age was 33 years. The COB was available for 1,381 (61.0%) patients; 1,157 (83.8%) were born in 89 countries outside the United Kingdom (Appendix Table). The population included representatives from all regions of the world (20).
VNTR Data as Phylogenic Markers
Figure 1
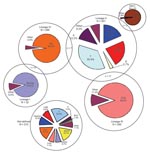
The 22 MIRU-VNTR genotypes, generated for 2,261 isolates, resulted in 1,434 VNTR types representing the minimum number of independent strains within this population. Each type was designated an MTBC lineage on the basis of the VNTR types (12) (Figure 1). Where these lineages were ambiguous (n = 49), discordant to those suggested by spoligotype (n = 58), or not defined (n = 210), SNP analysis was performed to resolve these differences (n = 317). In all cases, the SNP analysis resolved the ambiguous VNTR lineage calling as 1 of the alternatives producing the ambiguity. The SNP-defined lineage of strains discordant between the spoligotype and VNTR agreed with the VNTR call in 74.0% of cases. Finally, among the strains for which the VNTR was unable to define any lineage, there was 94.0% agreement between the SNP and spoligotype-defined lineage (Table 1). All strains identified as M. africanum were placed in the nondefined group and had the SNP-1 genotype.
Spoligotyping gave a lineage that was confirmed by an independent marker (VNTR or SNP) in 96.3% of isolates. VNTR gave an unambiguous lineage in 77.9% of strains; of these, 99% were confirmed by an independent marker (spoligo or SNP). Allelic variants were sought at each VNTR locus that best described each spoligofamily; those giving the highest sensitivities and specificities are shown in Table 2. The highest sensitivities were seen in the LAM 1, LAM 10, and Beijing families, which suggests their highly clonal and homogeneous nature. Several allelic variants showed strong associations with spoligo families, with >5 copies at ETR-A, >2 copies at MIRU24, and >3 copies at ETR-B associated with EAI and M. bovis (RR 2.99, 95% confidence interval [CI] 2.51–3.56; RR 6.29, 95% CI 4.87–8.12; and RR 3.21, 95% CI 2.63–3.93, respectively), >3 copies at MIRU4 and 2 copies at MIRU26 with EAI (RR 2.31, 95% CI 1.98–2.70; and RR 12.8, 95% CI 8.41–17.90, respectively), and 4 copies at MIRU23 with M. africanum and M. bovis (RR 220.3, 95% CI 82.07–591.50).
The presence of 2 copies at MIRU24 appears to be a good marker for EAI M. tuberculosis and non–M. tuberculosis members of the MTBC. This marker (number of copies in the locus MIRU24) was investigated in this population by using the occurrence of the deletions RD9 and TbD1, which have previously been used as markers to distinguish these groups (Table 3). All 41 isolates identified as M. africanum by spoligotype were also analyzed in this manner, 11 of which contained a single copy of MIRU24; 296 M. tuberculosis isolates containing single and double copies of MIRU24 were analyzed as controls.
Figure 2
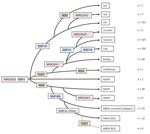
Figure 2. Maximum-parsimony tree constructed based on 3 independent sets of markers: large sequence polymorphisms (LSPs), single nucleotide polymorphisms (SNPs), and number of repeats in the locus 24 using the following assumptions: 1)...
The deletion TbD1 was present in all EuroAm, CAS, and Beijing strains examined as well as some other M. tuberculosis isolates and absent from all M. africanum isolates. The deletion RD9 was present in all M. bovis strains as well as some EAI and most M. africanum strains but absent from all other strains. Both deletions were absent from most EAI and some M. africanum strains (Table 3). Absence of RD9 deletion and 2 copies in MIRU24 was strongly associated with EAI spoligotype (RR 15.1, 95% CI 9.49–23.89). MTBC strains with the RD9 intact and 2 copies in MIRU24 included both M. bovis and M. africanum spoligotypes, whereas strains with the RD9 intact and 1 copy in MIRU24 formed a specific group of M. africanum originating presumably from the Indian subcontinent. Using this data, and the SNP 1-MB and the MIRU24 enumeration data, we constructed a maximum-parsimony tree as shown in Figure 2.
Associations between Phylogenic Groups and Phenotype
Strong associations were seen between patient’s country of origin and the spoligo family of the isolate (Appendix Table): CAS and EAI families dominated in patients born on ISC (RR 2.4, 95% CI 2.02–2.74) as did Beijing and EAI families in patients born in Southeast Asia (RR 4.8, 95% CI 2.70–8.54). EAI families were seen in 80.4% of isolates from patients born in East Africa and the ISC. The M. africanum family dominated in patients born in West Africa (RR 3.67, 95% CI 1.52–6.50). In contrast, LAM and Haarlem isolates were infrequently seen in patients born on the ISC (4.5% and 5.4%) and Southeast Asia (3.4% and 6.9%). T family isolates, one of the genetic groups determined by spoligotyping, were distributed evenly across all regions except Southeast Asia, where they were infrequently seen. No association between lineage or spoligo family and pulmonary versus extrapulmonary site was seen in the present study.
Susceptibility to rifampin, isoniazid, ethambutol, streptomycin, and pyrazinamide was evident for 98.9% (2,236) of the isolates. Of these isolates, 84.3% were sensitive to all, 8.2% were isoniazid resistant, 5.4% streptomycin resistant, 1.5% rifampin resistant, 0.7% ethambutol resistant, 0.5% pyrazinamide resistant, and 1.2% multidrug resistant.
Associations between spoligotype families and drug resistance of MTBC strains were analyzed by determining the minimum number of independent clones and the minimum number of resistance acquisition events within this population. VNTR15 cluster analysis was performed on all isolates (n = 2,261) to identify a single representative of each unique genotype. This analysis resulted in 1,166 unique types.
When isolates shared a genotype but differed in susceptibility to a given drug, resistant and sensitive isolates were analyzed because the resistant isolate must have undergone a genetic event and acquired a unique genotype. When genotypes for loci associated with isoniazid and rifampin resistance had been determined and >1 type was present in a cluster, 1 of each type was included. Where members of a cluster and its nearest neighbor were resistant, this was considered as a single acquisition event and only a single member was included. The resulting numbers divided between spoligotype families are shown in Table 4.
The M. bovis BCG family was associated with pyrazinamide (p<0.0001) and ethambutol resistance (p = 0.0009). Beijing family strains were associated with multidrug resistance (p = 0.0001), isoniazid (p = 0.0019), and rifampin (p = 0.0027) resistance. Associations were seen between streptomycin resistance and the Beijing family (p = 0.0008) and between pyrazinamide (p = 0.0079) and streptomycin (p = 0.008) resistance and the LAM1 family.
Several approaches have been used to study the global diversity of MTBC. One approach is to construct a global sample of isolates from reference collections around the world (19,30,31). In this instance, the degree of confidence as to geographic origin of an isolate is high, but bias occurs 1) where variety is limited to sites with which investigators have contact and 2) sites with high TB transmission, which often lack adequate facilities for bacteriologic culture. A second approach is to study isolates derived from a population at a single geographic location whose members have diverse geographic origins throughout the world (19,30,31). In this instance, where country of birth data are used to indicate the geographic origin of an isolate, the degree of confidence in this data may be lower, but MTBC isolates can be sampled at a wider range of geographic locations particularly from high TB incidence areas that have poor bacteriologic isolation facilities. Furthermore, additional data such as antimicrobial drug susceptibility and site of infection, useful for association studies, are retained and the quality of the data is ensured.
London is a cosmopolitan city where up to 30% of the population is foreign born (www.neighbourhood.statistics.gov.uk), among whom 75% of TB cases are seen (19; HPA update, www.hpa.org.uk); a similar situation has been reported in New York and Paris (19,30,31), although London TB notification rates (44.8 cases/100,000 population in 2006) are generally higher than those for other high-income cities. We believe that London provides a suitable setting for studying global MTBC diversity because our study shows that TB patients came from 89 different countries of origin, representing all regions of the world (20), including areas that the World Health Organization has defined as having a high incidence of TB. The bacterial diversity within this population is shown by the presence of all the main spoligofamilies, although not all lineages are equally represented. Our study shows a disproportionate representation of patients from different regions; relatively small numbers were from the Americas.
Recent advances in molecular genotyping and comparative genomics have demonstrated that the level of genetic variation in the MTBC may have been substantially underestimated. Rapidly evolving genomic regions such as VNTR and the direct repeat region have been exploited for epidemiologic studies, whereas irreversible events recorded by SNPs and LSPs are of phylogenetic value (3,5–7). Associations between polymorphisms in rapidly evolving genomic regions (VNTR or direct repeat region) and the SNP and LSP markers have been described (6,12,13,28,32–34). If the nature of these relationships could be clearly defined, large studies could be performed by investigating databases containing routine VNTR data.
Where lineages indicated by SNP and LSP analysis are congruent with spoligotype family names, we have retained these (as for CAS, EAI, Beijing, M. africanum and M. bovis); for the lineages containing LAM, Haarlem, X,T, and S spoligo families, we have used the lineage designation EuroAm as suggested elsewhere (6). We have previously reported 10 VNTR loci (ETR A,B,C; MIRU10,16,23, 24, 26,39,40) (12) capable of differentiating the MTBC into 4 lineages (I–IV) and M. bovis (7).
VNTR analysis showed that 1,174 (81.9%) of 1,434 independent strains could be grouped unambiguously into 5 lineages. When the remainder were grouped by using the SNP analysis, a good correlation was seen between lineage and spoligotype family or group of families (Figures 1, 2).
Discrepancies between lineage and spoligo family mainly resulted from limitations imposed by the family designation software, choice of genetic targets analyzed, or overlapping rules defining some spoligo families. Strains belonging to families 33–36 and EAI 1 appeared in multiple lineages. These spoligotype families were designated as low probability, which suggests that the model spoligotype was detecting unrelated events in different families. In rare cases, discrepancies will be seen where genetic events converge to give identical types in unrelated strains. In the present study this can be seen when multiple lineages are indicated by VNTR or spoligotypes.
Discrepancies will also occur where the VNTR/SNP system fails to distinguish between spoligotype families. The most striking of these are the strains identified as M. africanum by spoligotype but as the Beijing lineage because of the presence of SNP1. We resolved this problem by constructing a maximum-parsimony tree (Figure 2) using the 5 SNP, LSP, and MIRU24 repeat numbers. The MIRU24 repeat numbers appear to play a phylogenetic role, as shown in this study (Table 3) and previous studies (13,15) in which >2 repeats are markers for EAI2–EAI5 (but not EAI1), M. tuberculosis, and M. bovis strains. In its construction, we made the assumptions that SNPs mark irreversible unique events and that VNTR loci can acquire and lose repeats. A BCG strain isolated from a patient from London (not included in this study) contained the TbD1 deletion, demonstrating clearly that these deletion events are not unique. Therefore, the assumption that LSPs are infrequent irreversible events was made. The strains in this study are of human origin and therefore are mainly M. tuberculosis and M. africanum, hence the focus of the phylogenetic scenario. The tree shown here is concordant with previous scenarios (3,6) differing only in the diversity seen in strains identified as M. africanum. All these strains contained SNP1 and were identified on the basis of the loss of spoliogotype spacers 8, 9, and 39 but contained either 1 or 2 copies of MIRU24 and the presence and absence of RD9, resulting in 3 types. The absence of the TBD1 deletion distinguishes M. africanum strains from Beijing strains.
EAI strains may represent the ancestral MTBC type (6,15). The data presented here suggest that M. africanum competes for this distinction. The types containing 2 copies of MIRU24, with and without RD9 originate exclusively from West Africa, suggest that these may be indigenous to this region. M. africanum species have traditionally been phenotypically subdivided into 2 subgroups, Type 1 (West African) and Type 2 (East African) (34). Recent genetic analysis suggests that M. africanum Type 2 (East African) is a phenotypic variant of M. tuberculosis and relatively distant from M. africanum Type 1 (West African), which is characterized by a deleted RD9, an intact TbD1 region, and specific SNPs in katG and gyrA genes (35,36). Our data suggest a more complex phylogeny of M. africanum Type 1 (West African). This phylogeny is complicated further by strains with a deleted RD9 and a single copy of MIRU24 originating predominantly from the Indian subcontinent.
The VNTR numbers seen within each spoligo family are shown in Table 3. From these data, lineage-dependent VNTR locus plasticity can be seen. This plasticity ranges from 7/15 loci showing variation within the CAS to 14/15 showing variation in the EuroAm lineage. VNTR loci such as MIRU10 and 16 show variation across all families, whereas MIRU27 shows variation in CAS alone. The distribution of repeat numbers at each locus within each lineage suggests the variation seen has arisen by stepwise mutations of a lineage founder strain. It is likely that the VNTR profiles used to predict spoligotype family at the highest specificity (Table 2) represent this type.
Using country of birth as a surrogate for geographic origin of an infecting strain, we saw strong associations with the lineage/spoligo family of isolates (Appendix Table). The data here confirm published data that Beijing strains were associated with patients originating from Southeast Asia; EAI with patients from Southeast Asia, Indian subcontinent, and East Africa; CAS with patients from the Indian subcontinent; and EuroAm with a global distribution of patients (7,32). This global geographic structuring may explain the apparent geographic variation in efficacy of the M. bovis BCG vaccine.
It has been long questioned whether there is an association between site and progression of infection and bacterial genotype; some evidence supports this association (37,38). Our study showed no association between lineage or spoligo family and site of infection.
That the M. bovis family was associated with pyrazinamide resistance would be expected because resistance is a defining characteristic for most of the group (although not for M. bovis subsp. caprae). Beijing family isolates were associated with multidrug resistance and streptomycin resistance. The association with multidrug resistance has been reported (8), but the evidence presented here is particularly compelling, given that all strains used in the analysis were individual types. The value of this approach was demonstrated by analyzing LAM10 isolates, a family to which a highly successful clone of isoniazid-resistant M. tuberculosis responsible for >250 cases in northern London (38) belongs. Eight isolates were identified in this study. When all isolates belonging to this group were analyzed, LAM10 was strongly associated with isoniazid resistance (p<0.00001), but when a single representative of each cluster was used this association disappeared. The Beijing lineage would appear to have a predisposition toward the acquisition of drug resistance rather than the drug-resistant clones being transmitted more frequently. The extent of the geographic regions used in the association study make it unlikely that this predisposition is entirely due to local TB control and treatment practices.
Dr Brown is a clinical scientist at the UK Health Protection Agency National Reference Mycobacterium Laboratory and honorary senior lecturer at Barts and The London School of Medicine, Queen Mary, University of London, UK. He has a particular interest in clinical microbiology and molecular biology of human respiratory pathogens, especially tuberculosis.
Acknowledgments
We thank staff members at all participating hospitals and laboratories for their valuable help and support. We especially thank Carmel Prendergast for providing bacterial isolates.
This research was supported by the UK Department of Health grant Genotyping of Mycobacterium tuberculosis in London.
References
- World Health Organization. Global tuberculosis control: surveillance, planning, financing. WHO report 2007 (WHO/HTM/TB/2007.376) [cited 2009 Jul 10]. http://www.who.int/tb/publications/global_report/2007/pdf/full.pdf
- Sreevatsan S, Pan X, Stockbauer KE, Connell ND, Kreiswirth BN, Whittam TS, Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci U S A. 1997;94:9869–74. DOIPubMedGoogle Scholar
- Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A. 2002;99:3684–9. DOIPubMedGoogle Scholar
- Alland D, Lacher DW, Hazbon MH, Motiwala AS, Qi W, Fleischmann RD, Role of large sequence polymorphisms (LSPs) in generating genomic diversity among clinical isolates of Mycobacterium tuberculosis and the utility of LSPs in phylogenetic analysis. J Clin Microbiol. 2007;45:39–46. DOIPubMedGoogle Scholar
- Gutacker MM, Mathema B, Soini H, Shashkina E, Kreiswirth BN, Graviss EA, Single-nucleotide polymorphism-based population genetic analysis of Mycobacterium tuberculosis strains from 4 geographic sites. J Infect Dis. 2006;193:121–8. DOIPubMedGoogle Scholar
- Gagneux S, Small PM. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. 2007;7:328–37. DOIPubMedGoogle Scholar
- Baker L, Brown T, Maiden MC, Drobniewski F. Silent nucleotide polymorphisms and a phylogeny for Mycobacterium tuberculosis. Emerg Infect Dis. 2004;10:1568–77.PubMedGoogle Scholar
- Drobniewski F, Balabanova Y, Nikolayevsky V, Ruddy M, Kuznetzov S, Zakharova S, Drug-resistant tuberculosis, clinical virulence, and the dominance of the Beijing strain family in Russia. JAMA. 2005;293:2726–31. DOIPubMedGoogle Scholar
- Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14.PubMedGoogle Scholar
- Vitol I, Driscoll J, Kreiswirth B, Kurepina N, Bennett KP. Identifying Mycobacterium tuberculosis complex strain families using spoligotypes. Infect Genet Evol. 2006;6:491–504. DOIPubMedGoogle Scholar
- Brudey K, Driscoll JR, Rigouts L, Prodinger WM, Gori A, Al-Hajoj SA, Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol. 2006;6:23. DOIPubMedGoogle Scholar
- Gibson A, Brown T, Baker L, Drobniewski F. Can 15-locus mycobacterial interspersed repetitive unit-variable-number tandem repeat analysis provide insight into the evolution of Mycobacterium tuberculosis? Appl Environ Microbiol. 2005;71:8207–13. DOIPubMedGoogle Scholar
- Ferdinand S, Valetudie G, Sola C, Rastogi N. Data mining of Mycobacterium tuberculosis complex genotyping results using mycobacterial interspersed repetitive units validates the clonal structure of spoligotyping-defined families. Res Microbiol. 2004;155:647–54. DOIPubMedGoogle Scholar
- Kremer K, Au BK, Yip PC, Skuce R, Supply P, Kam KM, Use of variable-number tandem-repeat typing to differentiate Mycobacterium tuberculosis Beijing family isolates from Hong Kong and comparison with IS6110 restriction fragment length polymorphism typing and spoligotyping. J Clin Microbiol. 2005;43:314–20. DOIPubMedGoogle Scholar
- Gutierrez MC, Ahmed N, Willery E, Narayanan S, Hasnain SE, Chauhan DS, Predominance of ancestral lineages of Mycobacterium tuberculosis in India. Emerg Infect Dis. 2006;12:1367–74.PubMedGoogle Scholar
- van Deutekom H, Supply P, de Haas PE, Willery E, Hoijng SP, Locht C, Molecular typing of Mycobacterium tuberculosis by mycobacterial interspersed repetitive unit-variable-number tandem repeat analysis, a more accurate method for identifying epidemiological links between patients with tuberculosis. J Clin Microbiol. 2005;43:4473–9. DOIPubMedGoogle Scholar
- Oelemann MC, Diel R, Vatin V, Haas W, Rusch-Gerdes S, Locht C, Assessment of an optimized mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing system combined with spoligotyping for population-based molecular epidemiology studies of tuberculosis. J Clin Microbiol. 2007;45:691–7. DOIPubMedGoogle Scholar
- Durmaz R, Zozio T, Gunal S, Allix C, Fauville-Dufaux M, Rastogi N. Population-based molecular epidemiological study of tuberculosis in Malatya, Turkey. J Clin Microbiol. 2007;45:4027–35. DOIPubMedGoogle Scholar
- Anderson SR, Maguire H, Carless J. Tuberculosis in London: a decade and a half of no decline [corrected]. Thorax. 2007;62:162–7. DOIPubMedGoogle Scholar
- United Nations. Demographic Yearbook 2000. UN Document ST/ESA/STAT/SER.R/31 2000 [cited 2009 Jul 10]. http://www.un.org/esa/desa/desaNews/desa72.html
- Collins CH, Grange JM, Yates MD. Tuberculosis bacteriology organization and practice. Oxford (UK): Butterworth-Heinemann; 1997.
- Brown TJ, Herrera-Leon L, Anthony RM, Drobniewski FA. The use of macroarrays for the identification of MDR Mycobacterium tuberculosis. J Microbiol Methods. 2006;65:294–300. DOIPubMedGoogle Scholar
- Kwara A, Schiro R, Cowan LS, Hyslop NE, Wiser MF, Roahen Harrison S, Evaluation of the epidemiologic utility of secondary typing methods for differentiation of Mycobacterium tuberculosis isolates. J Clin Microbiol. 2003;41:2683–5. DOIPubMedGoogle Scholar
- Gopaul KK, Brown TJ, Gibson AL, Yates MD, Drobniewski FA. Progression toward an improved DNA amplification-based typing technique in the study of Mycobacterium tuberculosis epidemiology. J Clin Microbiol. 2006;44:2492–8. DOIPubMedGoogle Scholar
- Nikolayevskyy V, Gopaul K, Balabanova Y, Brown T, Fedorin I, Drobniewski F. Differentiation of tuberculosis strains in a population with mainly Beijing-family strains. Emerg Infect Dis. 2006;12:1406–13.PubMedGoogle Scholar
- Velji P, Nikolayevskyy V, Brown T, Drobniewski F. Discriminatory ability of hypervariable variable number tandem repeat loci in population-based analysis of Mycobacterium tuberculosis strains, London, UK. Emerg Infect Dis. 2009;15:1609–16.PubMedGoogle Scholar
- Allix-Beguec C, Fauville-Dufaux M, Supply P. Three-year population-based evaluation of standardized mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2008;46:1398–406. DOIPubMedGoogle Scholar
- Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol. 1988;26:2465–6.PubMedGoogle Scholar
- New York City Department of Health and Mental Hygiene. 2003 TB Annual Summary [cited 2009 Jul 10]. http://www.nyc.gov/html/doh/downloads/pdf/tb/tb2003.pdf
- Che D, Bitar D, Desenclos JC. Epidemiology of tuberculosis in France [in French]. Presse Med. 2006;35:1725–32. DOIPubMedGoogle Scholar
- Gagneux S, DeRiemer K, Van T, Kato Maeda M, de Jong BC, Narayanan S, Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:2869–73. DOIPubMedGoogle Scholar
- Nikolayevskyy VV, Brown TJ, Bazhora YI, Asmolov AA, Balabanova YM, Drobniewski FA. Molecular epidemiology and prevalence of mutations conferring rifampicin and isoniazid resistance in Mycobacterium tuberculosis strains from the southern Ukraine. Clin Microbiol Infect. 2007;13:129–38. DOIPubMedGoogle Scholar
- Mathema B, Kurepina NE, Bifani PJ, Kreiswirth BN. Molecular epidemiology of tuberculosis: current insights. Clin Microbiol Rev. 2006;19:658–85. DOIPubMedGoogle Scholar
- Collins CH, Yates MD. Mycobacterium africanum and the ‘African’ tubercle bacilli. Med Lab Sci. 1984;41:410–3.PubMedGoogle Scholar
- Niemann S, Kubica T, Bange FC, Adjei O, Browne EN, Chinbuah MA, The species Mycobacterium africanum in the light of new molecular markers. J Clin Microbiol. 2004;42:3958–62. DOIPubMedGoogle Scholar
- Sola C, Rastogi N, Gutierrez MC, Vincent V, Brosch R, Parsons L. Is Mycobacterium africanum subtype II (Uganda I and Uganda II) a genetically well-defined subspecies of the Mycobacterium tuberculosis complex? J Clin Microbiol. 2003;41:1345–6. DOIPubMedGoogle Scholar
- Caws M, Thwaites G, Dunstan S, Hawn TR, Lan NT, Thuong NT, The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog. 2008;4:e1000034. DOIPubMedGoogle Scholar
- Ruddy MC, Davies AP, Yates MD, Yates S, Balasegaram S, Drabu Y, Outbreak of isoniazid resistant tuberculosis in north London. Thorax. 2004;59:279–85. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 16, Number 2—February 2010
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Francis Drobniewski, UK Health Protection Agency, National Mycobacterium Reference Laboratory, Cfi, Clinical TB and HIV Research Group, Queen Mary, University of London, 2 Newark St, London E1 2AT, UK
Top