Volume 18, Number 1—January 2012
Letter
Hantavirus in Bat, Sierra Leone
Figure
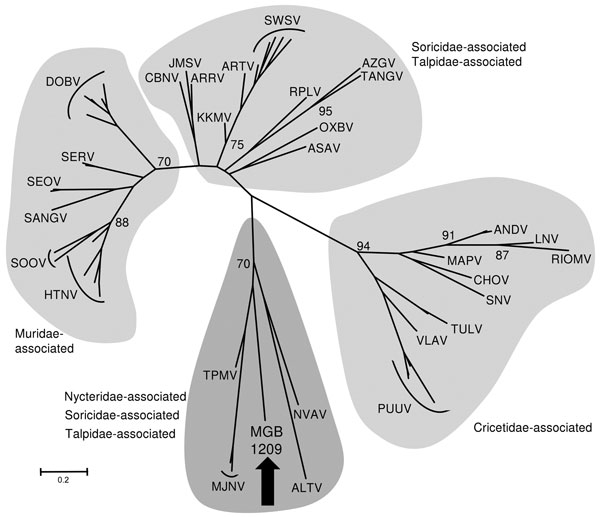
Figure. Maximum-likelihood phylogenetic tree of MGB/1209 virus based on partial large segment sequence (414 nt) and showing the phylogenetic placement of the novel sequence from Nycteris spp. bat compared with hantaviruses associated (i) with shrews and moles: Altai virus (ALTV), Artybash virus (ARTV), Asama virus (ASAV), Ash River virus (ARRV), Azagny virus (AZGV), Camp Ripley virus (RPLV), Cao Bang virus (CBNV), Imjin virus (MJNV), Jemez Springs virus (JMSV), Kenkeme virus (KKMV), Nova virus (NVAV), Oxbow virus (OXBV), Seewis virus (SWSV), Tanganya virus (TGNV), Thottapalayam virus (TPMV), and (ii) with rodents: Andes virus (ANDV), Choclo virus (CHOV), Dobrava-Belgrade virus (DOBV), Hantaan virus (HTNV), Laguna Negra virus (LNV), Maporal virus (MAPV), Puumala virus (PUUV), Rio Mamore virus (RIOMV), Sangassou virus (SANGV), Seoul virus (SEOV), Serang virus (SERV), Sin Nombre virus (SNV), Soochong virus (SOOV), Tula virus (TULV), Vladivostok virus (VLAV). The list of the accession numbers used in the analysis is available from the authors upon request. The tree was computed by using MEGA5 (http://www.megasoftware.net). The Tamura 3-parameter model with gamma-distributed rate heterogeneity and a proportion of invariant sites (T92 +G + I) was selected as the best fit evolutionary model according to the Baeysian information criterion calculated with MEGA5. The values at the tree branches are the bootstrap support values calculated from 500 replicates. Scale bar indicates an evolutionary distance of 0.2 substitutions per position in the sequence. The gray areas indicate association of hantaviruses with reservoir host families. The MGB/1209 partial sequence of the large genomic segment was deposited in GenBank under accession no. JN037851.
1These authors contributed equally to this article.