Volume 20, Number 4—April 2014
Letter
Pandemic Vibrio parahaemolyticus, Maryland, USA, 2012
To the Editor: Since 1996, an increasing number of infections caused by Vibrio parahaemolyticus strains belonging to a pandemic clonal complex (CC), CC3, typically O3:K6, have been observed worldwide (1–3); most of these strains are sequence type (ST) 3. In the summer of 1998, outbreaks linked to O3:K6 occurred in Galveston Bay, Texas, and Oyster Bay, New York, USA; the illnesses were associated with oyster consumption (4). Strains belonging to CC36 are the leading cause of V. parahaemolyticus infections in the United States. These strains are endemic to the West Coast (2) and have been historically linked to outbreak-associated V. parahaemolyticus infections caused by consumption of raw oysters harvested from the region (5).
In August 2012, a V. parahaemolyticus outbreak involving 6 persons occurred in Maryland, USA. The patients (members of 2 dining parties) had eaten in the same restaurant on the same day; raw and cooked seafood was served at the restaurant. Party A comprised 4 diners, of whom 2 had laboratory-confirmed illness and 2 were probable case-patients. Party B comprised 2 diners, of whom 1 had laboratory-confirmed illness and 1 was a probable case-patient. Probable case-patients were epidemiologically linked to confirmed case-patients, but V. parahaemolyticus was not detected in their stool samples. The epidemiologic investigation did not conclusively identify the specific food responsible for the outbreak. The affected diners had not eaten oysters, lobster, or mussels, but they had eaten cooked clams, fish, crab, and shrimp. Because the patients had not eaten oysters, a traceback investigation was not conducted. The outbreak possibly was caused by cross-contamination during food preparation. No other cases were reported from this restaurant or the surrounding area.
V. parahaemolyticus was isolated from stool samples of 3 of the patients. The isolates were characterized by real-time PCR for virulence-related genes (tdh and trh). All 3 isolates were tdh positive and lacked the trh gene. Pulsed-field gel electrophoresis (PFGE) was run, using Sfil and Notl; the resulting K16S12.0138 (Sfil) and K16N11.0143 (Notl) patterns were indistinguishable. The PFGE pattern combination was queried against combination entries made in PulseNet (www.cdc.gov/pulsenet/) during February 4, 2010–April 16, 2013, and found to be indistinguishable from other clinical entries (Technical Appendix 1, Table). This PFGE pattern combination has been seen 25 times; all patterns were for strains from humans (N. Facundo, pers. comm.). In 2012, this PFGE pattern combination was observed in 3 US states—California (6 cases), Arizona (6 cases), and Texas (5 cases)—but those isolates were not further tested (S.G. Stroika, pers. comm.), suggesting that other cases of pandemic V. parahaemolyticus infections have occurred in the United States but were not identified as being caused by pandemic clones.
Figure
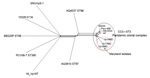
Figure. . Neighbor-Net graph generated with the Bacterial Isolate Genome Sequence Database genome (BIGSdb) comparator tool implemented within the Vibrio parahaemolyticus MLST database (http://pubmlst.org/vparahaemolyticus) (7,8) using 2,613...
The whole genomes of the 3 Maryland strains were sequenced by using the Ion Torrent personal genome machine (Life Technologies, Grand Island, NY, USA); in silico multilocus sequence typing (MLST) (2) showed that the isolates were all ST3, the most common ST belonging to CC3. Bioinformatic analysis of the whole genomes was conducted with the Bacterial Isolate Genome Sequence Database (6) genome comparator tool available within the V. parahaemolyticus MLST database (http://pubmlst.org/vparahaemolyticus) (7,8). Results confirmed that these outbreak isolates were linked to the O3:K6 pandemic clone of V. parahaemolyticus (Figure). We identified 2,613 variable loci in this analysis by using as reference genome the prototype pandemic V. parahaemolyticus clonal strain RIMD221633 (available from GenBank, www.ncbi.nlm.nih.gov/genome/?term=vibrioparahaemolyticus) (10). Differences in variable loci and the absence of certain genes indicated that, although indistinguishable by MLST and PFGE, these strains are easily differentiated from RIMD2210633 (Technical Appendix 1). The draft genome sequences for the 3 strains are available at the V. parahaemolyticus MLST database (identification nos. 1187 [Vp16MD], 1188 [Vp17MD], and 1189 [Vp18MD]).
V. parahaemolyticus strains belonging to the pandemic CC have caused thousands of infections and a V. parahaemolyticus pandemic (3). Foodborne illnesses caused by pandemic V. parahaemolyticus are uncommonly reported in the United States. In Maryland, 12 and 21 cases of V. parahaemolyticus–associated gastroenteritis were reported in 2012 and 2013, respectively. We report that the pandemic CC was still causing US outbreaks as recently as August 2012. It is possible that complete availability of PFGE patterns during the outbreaks (online Technical Appendix 1) could have provided additional insight into the scope of the outbreak and implicated food sources. The application of rapid, whole-genome sequencing technology aided our discovery that the Maryland outbreak strains were part of the pandemic CC and likely related to V. parahaemolyticus strains that shared common PFGE patterns and that were reported as the cause of illnesses in several states around the same time as the Maryland outbreak.
The presence of this virulent V. parahaemolyticus strain in Maryland is an ongoing public health concern, requiring continued microbiological surveillance. This pandemic strain also indicates the need for establishing a V. parahaemolyticus genome database that is accessible worldwide. Such a database would enable improved tracking and faster responses to emergent and dangerous pandemic clonal strains.
References
- González-Escalona N, Cachicas V, Acevedo C, Rioseco ML, Vergara JA, Cabello F, Vibrio parahaemolyticus diarrhea, Chile, 1998 and 2004. Emerg Infect Dis. 2005;11:129–31. DOIPubMedGoogle Scholar
- González-Escalona N, Martinez-Urtaza J, Romero J, Espejo RT, Jaykus LA, DePaola A. Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J Bacteriol. 2008;190:2831–40. DOIPubMedGoogle Scholar
- Nair GB, Ramamurthy T, Bhattacharya SK, Dutta B, Takeda Y, Sack DA. Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin Microbiol Rev. 2007;20:39–48. DOIPubMedGoogle Scholar
- DePaola A, Kaysner CA, Bowers J, Cook DW. Environmental investigations of Vibrio parahaemolyticus in oysters after outbreaks in Washington, Texas, and New York (1997 and 1998). Appl Environ Microbiol. 2000;66:4649–54. DOIPubMedGoogle Scholar
- Abbott SL, Powers C, Kaysner CA, Takeda Y, Ishibashi M, Joseph SW, Emergence of a restricted bioserovar of Vibrio parahaemolyticus as the predominant cause of vibrio-associated gastroenteritis on the West Coast of the United States and Mexico. J Clin Microbiol. 1989;27:2891–3 .PubMedGoogle Scholar
- Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. DOIPubMedGoogle Scholar
- Jolley KA, Hill DM, Bratcher HB, Harrison OB, Feavers IM, Parkhill J, Resolution of a meningococcal disease outbreak from whole-genome sequence data with rapid Web-based analysis methods. J Clin Microbiol. 2012;50:3046–53. DOIPubMedGoogle Scholar
- Jolley KA, Maiden MC. Automated extraction of typing information for bacterial pathogens from whole genome sequence data: Neisseria meningitidis as an exemplar. Euro Surveill. 2013;18:20379 .PubMedGoogle Scholar
- Bryant D, Moulton V. Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol. 2004;21:255–65. DOIPubMedGoogle Scholar
- Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Genome sequence of Vibrio parahaemolyticus: a pathogenic mechanism distinct from that of V. cholerae. Lancet. 2003;361:743–9. DOIPubMedGoogle Scholar
Figure
Cite This ArticleRelated Links
Table of Contents – Volume 20, Number 4—April 2014
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Narjol Gonzalez-Escalona, Food and Drug Administration, Center for Food and Applied Nutrition, 5100 Paint Branch Pkwy, College Park, MD 20740, USANarjol Gonzalez-Escalona, Food and Drug Administration, Center for Food and Applied Nutrition, 5100 Paint Branch Pkwy, College Park, MD 20740, USA
Top