Volume 21, Number 12—December 2015
Letter
Emerging Rabbit Hemorrhagic Disease Virus 2 (RHDVb), Australia
Figure
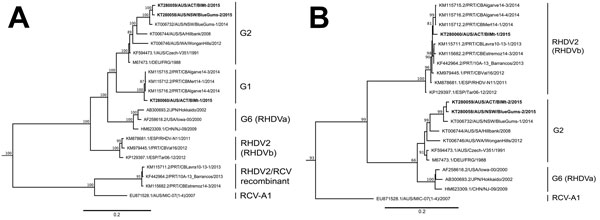
Figure. Maximum-likelihood phylogenetic analysis of the nonstructural protein genes (A) and the capsid gene (B) of rabbit hemorrhagic disease virus (RHDV) sequences. The 3 recent Australian field isolates sequenced for this study (indicated in bold) were aligned with representative RHDV and Australian rabbit calicivirus (RCV-A1) sequences from GenBank (accession numbers indicated in taxa names). Phylogenetic analysis was conducted separately for both the nonstructural genes (panel A) and the capsid gene (panel B). Phylogenies were rooted by using an early European brown hare syndrome virus strain (not shown). Statistical support for individual nodes was estimated from 1,000 bootstrap replicates with values shown for only those nodes where the bootstrap support was ≥70% (and all major nodes). Phylogenies were constructed by using the general time reversible plus gamma model of nucleotide substitution, as determined in jModelTest, by using PhyML(as available in Geneious version 8.1.5; Biomatters Limited, Auckland, New Zealand). Scale bars are proportional to the number of nucleotide substitutions per site.
1These first authors contributed equally to this article.