Volume 23, Number 9—September 2017
Research
Protective Effect of Val129-PrP against Bovine Spongiform Encephalopathy but not Variant Creutzfeldt-Jakob Disease
Abstract
Bovine spongiform encephalopathy (BSE) is the only known zoonotic prion that causes variant Creutzfeldt-Jakob disease (vCJD) in humans. The major risk determinant for this disease is the polymorphic codon 129 of the human prion protein (Hu-PrP), where either methionine (Met129) or valine (Val129) can be encoded. To date, all clinical and neuropathologically confirmed vCJD cases have been Met129 homozygous, with the exception of 1 recently reported Met/Val heterozygous case. Here, we found that transgenic mice homozygous for Val129 Hu-PrP show severely restricted propagation of the BSE prion strain, but this constraint can be partially overcome by adaptation of the BSE agent to the Met129 Hu-PrP. In addition, the transmission of vCJD to transgenic mice homozygous for Val129 Hu-PrP resulted in a prion with distinct strain features. These observations may indicate increased risk for vCJD secondary transmission in Val129 Hu-PrP–positive humans with the emergence of new strain features.
The presence of variant Creutzfeldt-Jakob disease (vCJD) is considered by strong epidemiologic, pathologic, and molecular evidence to be a likely consequence of human dietary exposure to the bovine spongiform encephalopathy (BSE) agent (1–3). Secondary vCJD infection has occurred through iatrogenic routes such as blood transfusion (4–7). The pathogenesis of these fatal transmissible spongiform encephalopathies (TSEs), called prion diseases, is associated with the accumulation of the abnormal isoform (PrPSc) of prion protein (PrP), which is converted from the normal cellular isoform (PrPC) (8). This conversion process involves a posttranslational conformational change of PrPC and PrPSc that can be distinguished biochemically from PrPC by its partial resistance to proteolysis and detergent insolubility (9,10).
The neuropathological features of vCJD are characterized by the presence of abundant florid PrP plaques and the propagation of type 4 disease-related PrPSc in the brain (1,11). Differences in the level of glycosylation, as well as in the size of protease-digested PrPSc, are widely used as surrogates of prion strain typing; 2 main classifications are recognized in the prion field (1,12). According to 1 of these classifications (1,13), type 4 PrPSc is characterized by a fragment size and glycoform ratio similar to that seen in BSE and BSE transmitted to several other species, with a predominance of the diglycosylated PrP glycoform (1,13–15).
Polymorphism at codon 129 of the human PrP gene (PRNP), where methionine (Met) or valine (Val) can be encoded, strongly affects susceptibility to human prion diseases (16–20). vCJD has only been neuropathologically confirmed in persons homozygous for Met at residue 129 of human PrP (21), with 1 exception of heterozygosity (Met/Val) at this codon (22). In addition, asymptomatic peripheral involvement in vCJD infection has been reported in 2 Met/Val129–positive persons (5,7). Retrospective studies of the prevalence of subclinical vCJD infection using appendectomy and tonsillectomy specimens in the United Kingdom described 6 appendixes that were positive for disease-associated prion protein in Val/Val129 persons (23–25). All of these human studies, in addition to the extremely prolonged and variable incubation periods seen in prion transmission experiments when crossing a species barrier, suggest that persons encoding any of the 3 human PrP codon 129 genotypes may be susceptible to vCJD, including secondary vCJD transmitted through blood transfusion, blood products, tissue and organ transplantation, and other iatrogenic routes.
Because only 1 definite case of heterozygous Met/Val129 vCJD and no homozygous Val129 cases have been described, it is unknown whether the clinicopathologic characteristics and biochemical properties of vCJD would appear in persons with these codon 129 genotypes. To gain insights into that question, vCJD/BSE transmission studies in which either humanized overexpressing or knock-in transgenic mice were used have been performed (2,26–30). However, some discrepancies in the transmission efficiency of vCJD to humanized knock-in transgenic mice can be found, depending on the origin of the mice and on the vCJD isolate (29,30). Previous studies in humanized overexpressing transgenic mice revealed that the 3 human PrP codon 129 genotypes can be infected with vCJD but show significant differences depending on the genotype. Moreover, mice with the Val/Val129 genotype were more susceptible to vCJD infection than expected but lack the neuropathological characteristics observed with Met/Met129 (2,26–28).
In this study, we evaluated the zoonotic potential of BSE and BSE adapted to different species by using transgenic mice overexpressing similar levels of human PrPC carrying Met/Met, Met/Val, or Val/Val at position 129 of human PrP. Furthermore, we used these models to re-evaluate the potential for human-to-human spread of vCJD, as well as the differential susceptibility and characteristics of the transmitted disease across the different PRNP codon 129 genotypes in humans.
Ethics Statement
We carried out animal experiments in strict accordance with the recommendations in the guidelines of the Code for Methods and Welfare Considerations in Behavioral Research with Animals (Directive 86/609EC and 2010/63/EU), and all efforts were made to minimize suffering. Experiments were approved by the Committee on the Ethics of Animal Experiments of the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (Madrid, Spain; permit nos. CEEA2012/024 and CEEA2009/004).
TSE Isolates
We used 11 isolates from different sources in this study (Table 1 [31–39]). For mouse inoculation, we prepared all isolates from brain tissues as 10% weight/volume (wt/vol) homogenates in 5% glucose. We performed second passages by inoculating transgenic mice with 10% (wt/vol) homogenates in 5% glucose of brains selected from passage 1.
Mouse Transmission Studies
We inoculated all isolates in 3 different transgenic mouse models: 1) HuPrP-Tg340-Met129 (TgMet129) mouse line expressing human Met129-PrPC variant (31); 2) HuPrP-Tg361-Val129 (TgVal129) mouse line expressing human Val129-PrPC variant (40); and 3) HuPrP-Tg351-Met/Val129 (TgMet/Val129) transgenic mouse line obtained by mating TgMet129 and TgVal129 mice (40). All of these transgenic lines show similar brain expression levels of PrPC (around 4-fold the level of expression in the human brain) on a mouse PrP null background. We performed additional inoculations in HuPrP-Tg362-Val129, a transgenic mouse line expressing 8-fold the level of PrPC expression in human brain (TgVal129 [8×]) (41). We performed subsequent bioassays for the detection of low-level propagation of infectious BSE and BSE-derived prions in BoPrP-Tg110 mice, which are highly susceptible to vCJD prions (42,43), probably caused by the trace-back phenomenon (30).
We anesthetized individually identified mice, 6–7 weeks of age, with isoflurane and inoculated them with a 2-mg equivalent of brain homogenate in the right parietal lobe by using a 25-gauge disposable hypodermic needle. We observed mice daily and assessed neurologic status 2 times per week. When progression of a TSE disease was evident or at the established experimental endpoint (700 days postinoculation [dpi]), we euthanized the animal for ethical reasons and performed necropsy, excising the brain. We then fixed part of the brain by immersion in neutral-buffered 10% formalin (4% 2-formaldehyde) and used the tissue for quantifying spongiform degeneration by histopathology. We froze the remaining tissue at −20°C and used it to determine the presence of disease-associated proteinase K (PK)–resistant PrP (PrPres) by Western blot.
In all cases, we calculated mouse survival time and disease attack rate for each isolate. We expressed survival times as mean ±SD of the dpi for all mice positive for PrPres. We defined the attack rate as the proportion of all inoculated mice whose samples tested positive for PrPres. We used brain homogenates from PrPres–positive mice, where available, for further passaging. When all mice were scored negative for PrPres on primary passage, we used PrPres-negative brain homogenates for second passage.
Western Blot
We homogenized frozen brain tissues (175 ± 20 mg) in 5% glucose in distilled water in grinding tubes (Bio-Rad, Hercules, CA, USA) adjusted to 10% (wt/vol) by using a TeSeE Precess 48TM homogenizer (Bio-Rad), according to the manufacturer’s instructions. We determined presence of PrPres in transgenic mouse brains by Western blot, using the reagents of the ELISA commercial test TeSeE (Bio-Rad). Based on a previously described protocol (31), we treated 10–100 μL of 10% wt/vol brain homogenates with proteinase K; the resulting samples were loaded in 12% Bis-Tris Gel (Criterion XT; Bio-Rad). We transferred proteins electrophoretically onto PVDF membranes (Millipore, Billerica, MA, USA), which were blocked overnight with 2% BSA blocking buffer (Sigma-Aldrich, St. Louis, MO, USA). For immunoblotting, we incubated with Sha 31 (44) monoclonal antibody (mAb) at a concentration of 1 µg/mL to identify the 145-WEDRYYRE-152 epitope of the human PrPC sequence. To detect immunocomplexes, we incubated the membranes for 1 h with horseradish peroxidase conjugated anti-mouse IgG (GE Healthcare Amersham Biosciences, Little Chalfont, UK). Immunoblots were developed with enhanced chemiluminiscence ECL Select (GE Healthcare Amersham Biosciences). Images were captured using the ChemiDoc WRS+ System (Bio-Rad) and processed using Image Lab 5.2.1 software (Bio-Rad).
Histopathological Analysis
We performed procedures for the histopathological analysis of mouse brains as previously described (45). We immediately fixed mouse brain samples in neutral-buffered 10% formalin (4% 2-formaldehyde) during necropsy and embedded the tissues in paraffin later. After deparaffinization, we stained 2 μm–thick tissue slices with hematoxylin and eosin and established lesion profiles by using published standard methods (46). We conducted paraffin-embedded tissue (PET) blots as previously described (47).
BSE Resistance in TgVal129 Mice
To evaluate the relative susceptibility of the 3 human PRNP codon 129 genotypes to BSE, we performed serial transmission studies in 3 transgenic mouse lines expressing human PrP. These mouse lines were homozygous for Met (TgMet129) or Val (TgVal129) at codon 129 of human PrP or were their F1 cross (TgMet/Val129). These mouse models expressed similar human PrP levels, ≈4-fold more than that seen in uninfected human brain tissue (40). We observed no clinical signs of prion disease or PrPres accumulation in control mice inoculated with TSE-free control brain homogenate. The 3 human transgenic mouse models were readily infected when inoculated with sporadic CJD (sCJD) (Table 2). The 2 sCJD cases used as inocula in this study were classified as Met129 type 1 (Hu-sCJD MM1) and Val129 type 2 (Hu-sCJD VV2) (12) on the basis of the patient´s PRNP genotype at codon 129 and the PrPres Western blot profiles of these samples.
We inoculated the 3 mouse models intracerebrally with a panel of BSE isolates from different species (cattle, pig, sheep, and goat; Table 2). As previously described in TgMet129 mice (31), we found a higher transmission efficiency adjudged by comparatively higher attack rates for BSE isolates previously passaged in other species than for cattle BSE, suggesting a strong transmission barrier to cattle BSE in these mice.
At completion of the first and second passages, none of the TgVal129 mice challenged with the different BSE isolates developed clinical disease, and no PrPres accumulation was found in their brains (Table 2). Because of intercurrent illnesses, the group of TgVal129 mice challenged with Ca-BSE2 was considerably reduced in size; however, the absence of transmission to TgVal129 mice challenged with a second BSE inocula, Ca-BSE0, reinforces this negative result. In addition, results of subsequent passage of brain homogenates from these mice to BoPrP-Tg110 mice were negative, ruling out the presence of subclinical infection, with the exception of TgVal129 mice inoculated with Go-BSE. For this isolate, 3 of 6 BoPrP-Tg110 mouse brains showed detectable PrPres and had a long incubation time of 427 ± 38 dpi, suggesting very low infectivity (Technical Appendix Table 1).
To confirm that the lack of susceptibility of TgVal129 mice to cattle BSE and to BSE previously adapted in different species was not caused by inadequate PrP substrate, we used the TgVal129 (8×) mouse line (41). However, even under these high human PrP expression level conditions, none of the inoculated TgVal129 (8×) mice showed any evidence of infection after challenge with the different BSE isolates (Technical Appendix Table 2). This result indicates that even an increase in the TgVal129 PrP expression level is not enough to allow transmission of BSE prions, irrespective of the species in which BSE has been previously passaged.
In a similar manner to that seen in the TgVal129 mice, we observed no clinical disease and no disease-associated PK-resistant PrP accumulation on first or second passage of the different BSE isolates in TgMet/Val129 mice. However, we did observe an exception in 1 TgMet/Val129 mouse inoculated with Go-BSE without clinical signs but with a positive score for brain PrPres that died at 476 dpi (Table 2). These findings support the interpretation that human-PrP Val129 polymorphism severely restricts propagation of the BSE prion strain independently of the species in which it had previously been adapted.
BSE Adaptation to the Human PrP Sequence
In parallel to the transmission experiments with the different BSE isolates, we also inoculated the 3 humanized transgenic mouse models with human brain material from 2 different cases of vCJD PrP-Met129 (Hu-vCJD1 and Hu-vCJD2). On first passage, 100% of the TgMet129 mice developed clinical disease in response to all inocula in the panel (Table 3). However, only the inoculum Hu-vCJD2 previously passaged in TgMet/Val129 mice caused clinical disease in the same heterozygous genotype upon serial passages; the rest of the inocula caused only subclinical infections in this genotype (Table 3).
Figure 1

Figure 1. Biochemical features of the protease-resistant prion protein (PrPres) detected in the brain of TgMet129, TgMet/Val129, and TgVal129 mice inoculated with vCJD. A) PrPres detected in TgMet129 (lanes 2 and 5), TgMet/Val129...
Figure 2
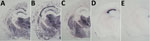
Figure 2. Protease-resistant prion protein distribution pattern in brains of prion protein humanized transgenic mice inoculated with variant Creutzfeldt-Jakob disease (vCJD) on second passage. A, B) TgMet129 mice inoculated with vCJD. C) TgMet/Val129...
The PrPres molecular profile (Figure 1, panel A, lanes 2, 3, and 5; Figure 1, panel B) and the PrPres distribution patterns on paraffin-embedded tissue (PET) blots in the mouse brains (Figure 2, panels A, B, C) were similar in both the TgMet129 and TgMet/Val129 mice, with or without clinical disease. However, we consistently observed a lower PrPres accumulation in TgMet/Val129 mice compared with TgMet homozygous animals, particularly in the hippocampus area, probably caused by a slower conversion rate of PrPSc in these animals with a half dose of PrP-Met129.
In sharp contrast, none of the TgVal129 mice challenged with the 2 vCJD primary inocula, Hu-vCJD1 and Hu-vCJD2, developed clinical disease and no PrPres accumulation was found in their brains after Western blot (WB) analysis (Table 3). However, subsequent passage of brain homogenates from TgVal129 mice inoculated with Hu-vCJD2 (that remained apparently uninfected) to BoPrP-Tg110 mice showed evidence of subclinical infection. These subpassages led to a mean incubation time of 371 ± 5 dpi and to propagation of PrPres that was detectable by WB in 100% of animals (Technical Appendix Table 1), showing a biochemical pattern indistinguishable from that of cattle BSE infection in this mouse model.
These results suggest that the adaptation of the BSE agent to human PrP sequence could favor its transmission to the polymorphic human PrP Val129 genotype. In this context, we passaged all isolates in TgMet129 mice before subsequent inoculation in TgVal129 mice. Although we did not observe clinical prion disease, the inoculated TgVal129 mice had an infection rate remarkably close to 100%, as assessed by the presence of brain PrPres at the end of the experiment (Table 3). We obtained similar results with the Hu-vCJD2 isolate after 1 passage in TgMet/Val129 mice and subsequent inoculation into TgVal129 mice (Table 3). These observations support the hypothesis that adaptation of BSE agent to the human-PrP Met129 amino-acid sequence promotes its transmission to human PrP Val129–expressing hosts.
vCJD Prions in TgVal129 Mice
Challenge of TgMet129 or TgMet/Val129 mice with vCJD prions resulted in faithful propagation of a typical PrPvCJD (also named type 4), characterized by low size fragments (19-kDa fragment for the aglycosyl band) and prominent diglycosylated species on WB (Figure 1, panel A, lanes 2, 3). These biochemical properties were accompanied by the key neuropathological hallmark of vCJD, the presence of abundant florid PrP plaques determined by immunohistochemical analysis of the brain (31) (data not shown).
In contrast, TgMet129–passaged vCJD-inoculated TgVal129 mice propagated a PrPSc with a WB signature that shared the same predominance of the diglycosylated glycoform seen in type 4 PrPSc but was distinguished by PK digestion products of greater molecular mass (Figure 1, panel A, lanes 4, 6), which closely resemble those seen in human type 2 PrPSc (Figure 1, panel A, lane 8). This differential biochemical pattern is associated with the presence of amyloid plaques restricted to the corpus callosum without a florid morphology. Moreover, we saw no specific vacuolar changes in the brains of these animals. PET blot analysis of these brains confirmed PrPSc deposition in corpus callosum and head of caudate nucleus in the brain of vCJD-inoculated TgVal129 mice (Figure 2, panels D, E). However, PrPSc deposition was quite limited in comparison with those observed in vCJD-inoculated TgMet129 (Figure 2, panels A, B) and TgMet/Val129 mice (Figure 2, panel C).
Figure 3
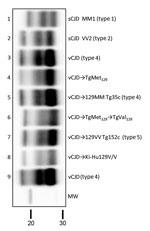
Figure 3. Biochemical comparison of brain protease-resistant prion protein (PrPres) detected in transgenic mice expressing prion protein Met129, and Val129 mice and inoculated with vCJD brain homogenate. Similar quantities of PrPres were loaded...
These results resemble those previously described in a different TgVal129 mouse line in which neuropathological and molecular features similar to those observed in our TgVal129 were characterized (2,27,28). To prove the same PrPres molecular profile identity between this previously characterized PrPSc (called type 5 PrPSc, vCJD→129VV Tg152c) and our vCJD-TgVal129 PrPSc, we performed a biochemical characterization by WB and found no molecular profile differences in PrPres from the various mouse lines (Figure 3, lanes 6 and 7). These particular molecular mass and glycoform profile characteristics seem to be a hallmark of vCJD transmission to human-PrP Val129, since these features were also found in a different human-PrP Val129 transgenic mouse line challenged with vCJD (vCJD→Ki-Hu129V/V) (26) (Figure 3, lane 8). These results, suggesting vCJD prion infection can result in the generation of distinct molecular and neuropathological phenotypes dependent on human-PrP polymorphic residue 129, are in accordance with those reported previously (2,28,46).
We report a detailed comparison of the transmission properties of BSE and vCJD prions among humanized transgenic mice with different PRNP codon 129 genotypes. Because a high expression level of PrP in transgenic mice directly influences prion disease susceptibility and incubation time, these transgenic mice have an advantage over knock-in mice for evaluating these features in the different human PrP genotypes. In addition, the 3 mouse models used in our study have equivalent PrP expression levels, making them suitable for studying comparative susceptibilities across the different PRNP codon 129 genotypes.
In previous reports, we demonstrated that Met129 homozygous individuals might be susceptible to a sheep or goat BSE agent to a higher degree than to cattle BSE and that these agents might transmit with molecular and neuropathological properties indistinguishable from those of vCJD (31). In this study, we wanted to extend these results to the other human PRNP genotypes: Met/Val129 and Val/Val129. We gained a different perspective when several BSE isolates adapted to different species inoculated in TgVal129 mice showed an apparent lack of transmission. In addition, almost all inoculated TgMet/Val129 mice did not transmit BSE; this finding supports the interpretation by Wadsworth et al. that human PrP Val129 severely restricts propagation of the BSE prion strain (27).
An unexpected result of this study was the finding that 1 BSE isolate from a goat (Ca-BSE/Go) was clinically transmitted to 1 of 10 TgMet/Val129 mice and subclinically transmitted to TgVal129 mice. This particular isolate is characterized by a high infectious titer (35) that could explain the potential for this inoculum to overcome the restriction on BSE prions to propagate in TgVal129 mice.
Although cattle BSE did not transmit to TgMet/Val129 mice directly, adaptation of the BSE agent to human PrP Met129 sequence and subsequent inoculation of the resultant vCJD prions to TgMet/Val129 mice produced a 100% attack rate. However, we did not detect clinical prion disease, supporting a slower rate of vCJD conversion compared with that among TgMet129 mice. This slow but potential conversion rate in TgMet/Val129 mice correlates well with the single vCJD case of a human carrying the PrP Met/Val129 genotype (22) and with the description of subclinical secondary transmissions through human vCJD–infected tissues (4–7,47).
TgVal129 mice challenged with Hu-vCJD2 did not produce detectable brain PrPres and clinical signs, in spite of the overexpression of HuPrP-Val129 and the use of the more efficient intracerebral route of infection. However, subclinical infection in these TgVal129 mice was demonstrated in BoPrP-Tg110 mice. These data suggest that PrP Val129 could sustain a very slow and limited vCJD conversion rate that is consistent with the detection of PrPres in tonsils and appendixes of asymptomatic PrP Val129 persons (23–25). Previous studies of other transgenic mice expressing PrP Val129 have also shown a low transmission efficiency of vCJD (2,27,30).
The fluctuating subclinical transmissibility of both vCJD inocula in TgVal129 mice (negative for Hu-vCJD1 and positive for Hu-vCJD2) might be caused by differences in prion titer between inocula. This assessment was strengthened after the transmission of both vCJDs to TgMet129 mice, in which a shorter incubation period was observed in animals inoculated with Hu-vCJD2. A certain variability in subclinical transmissibility and incubation time between different vCJD isolates is not uncommon, as has been previously reported (2,27,30), suggesting that a Val129 transmission barrier can only be overcome with highly infectious vCJD isolates.
The dramatic changes in the susceptibility of TgVal129 mice (Table 3) challenged with vCJD isolates first passaged in TgMet129 mice suggest an apparent increase in titer of both vCJD prion isolates; however, adaptation of the inocula to the new host mouse cannot be disregarded as being partly responsible for this increased susceptibility. We observed a 100% infection rate, but without clinical signs of prion disease. We observed similar transmission features when we passaged vCJD in TgMet/Val129 mice. In addition, the apparent PrPVal129 restricted propagation of cattle BSE and BSE from other species was completely abolished after its adaptation to human PrPMet129.
Although PrP overexpression and the inoculation route can affect transmission efficiency, our results and those previously reported in both overexpressing and knock-in transgenic mice (2,27,30) suggest that the Val129 PrP variant could sustain a very slow and limited vCJD conversion rate, and is unable to completely prevent vCJD transmission. Biochemical and neuropathological features of vCJD transmission to TgVal129 mice showed substantial differences compared to TgMet129 or TgMet/Val129 mice. Similar to previous reports (2,27,28,48), a type 5 PrPSc associated with very weak and diffuse PrP plaques without a florid morphology was the hallmark among these mice. In addition, our demonstration of previously unreported type 5 PrPSc in brain samples of vCJD-challenged knock-in Ki-Hu129V/V mice (30) establishes that the evolution of type 5 PrPSc associated with the transmission of vCJD prions to the Val129 genotype is not an artifact of PrP overexpression. This finding further reinforces the specific biochemical features of vCJD when transmitted to the human-PrP Val129 sequence.
Extrapolation of results from prion transmission studies based on transgenic mice has to be done with caution, especially when human susceptibility to prions is analyzed. However, our results clearly indicate that PrPVal129 individuals are highly resistant to transmission of cattle BSE or BSE passaged in other species. Also, PrPVal129 individuals might be susceptible to infection with human-passaged BSE (vCJD) prions, and the propagated agents might transmit with molecular and neuropathological properties distinguishable from those of type 4 PrPres. Although the resultant type 5 PrPSc shares the same fragment sizes as those of type 2 PrPSc, the 2 PrPSc types can be distinguished by the predominance of the diglycosylated glycoform associated with type 5 PrPSc. Overall, our results indicate that human Val129-PrP polymorphic variant is a strong molecular protector against BSE zoonotic transmission but fails to prevent human-to-human vCJD transmission. Because potential late-onset vCJD cases could appear in the population (49,50) these findings underline the need for continued investigation of all forms of human prion disease.
Dr. Fernández-Borges is a research scientist in the Prion Group at Centro de Investigación en Sanidad Animal–Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria, Madrid, Spain. Her research interests include prion strain characterization and evolution and the pathogenesis of prion diseases and their effects on human and animal health.
Acknowledgments
We thank Wilfred Goldmann for providing the goat-BSE material (gBSE-P12); Juan Piquer, Irene Prieto, Patricia Lorenzo, Ana Esteban, and Ana Villa for their technical assistance; and the staff of the Biosafety Level 3 animal facility and the Biosafety Office at the CISA-INIA (Valdeolmos-Madrid) for their excellent animal care and work.
This work was funded by the European Union projects CT-2006-36353 (GoatBSE), CT-2009-222887 (Priority), and 219235 ERANET EMIDA (GOAT-TSE-FREE); the UK Food Standards Agency grant M03043; the Alliance BioSecure Foundation grant FABS201403; and the Spanish Plan Nacional de I+D+I RTA2012-00004 and AGL2015-71764-REDT projects.
References
- Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–90. DOIPubMedGoogle Scholar
- Hill AF, Desbruslais M, Joiner S, Sidle KC, Gowland I, Collinge J, et al. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–50, 526. DOIPubMedGoogle Scholar
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. DOIPubMedGoogle Scholar
- Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–21. DOIPubMedGoogle Scholar
- Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527–9. DOIPubMedGoogle Scholar
- Wroe SJ, Pal S, Siddique D, Hyare H, Macfarlane R, Joiner S, et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet. 2006;368:2061–7. DOIPubMedGoogle Scholar
- Peden A, McCardle L, Head MW, Love S, Ward HJ, Cousens SN, et al. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia. 2010;16:296–304 . DOIPubMedGoogle Scholar
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;93:337–48. DOIPubMedGoogle Scholar
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–50. DOIPubMedGoogle Scholar
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–5. DOIPubMedGoogle Scholar
- Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–78. DOIPubMedGoogle Scholar
- Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, Budka H, et al. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain. 2003;126:1333–46. DOIPubMedGoogle Scholar
- Torres JM, Espinosa JC, Aguilar-Calvo P, Herva ME, Relaño-Ginés A, Villa-Diaz A, et al. Elements modulating the prion species barrier and its passage consequences. PLoS One. 2014;9:e89722. DOIPubMedGoogle Scholar
- Wadsworth JD, Collinge J. Update on human prion disease. Biochim Biophys Acta. 2007;1772:598–609.
- Palmer MS, Dryden AJ, Hughes JT, Collinge J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature. 1991;352:340–2. DOIPubMedGoogle Scholar
- Collinge J, Palmer MS, Dryden AJ. Genetic predisposition to iatrogenic Creutzfeldt-Jakob disease. Lancet. 1991;337:1441–2. DOIPubMedGoogle Scholar
- Mead S, Stumpf MP, Whitfield J, Beck JA, Poulter M, Campbell T, et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science. 2003;300:640–3. DOIPubMedGoogle Scholar
- Baker HE, Poulter M, Crow TJ, Frith CD, Lofthouse R, Ridley RM, et al. Aminoacid polymorphism in human prion protein and age at death in inherited prion disease. Lancet. 1991;337:1286. DOIPubMedGoogle Scholar
- Poulter M, Baker HF, Frith CD, Leach M, Lofthouse R, Ridley RM, et al. Inherited prion disease with 144 base pair gene insertion. 1. Genealogical and molecular studies. Brain. 1992;115:675–85. DOIPubMedGoogle Scholar
- Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol. 2000;47:575–82. DOIPubMedGoogle Scholar
- Bishop MT, Diack AB, Ritchie DL, Ironside JW, Will RG, Manson JC. Prion infectivity in the spleen of a PRNP heterozygous individual with subclinical variant Creutzfeldt-Jakob disease. Brain. 2013;136:1139–45. DOIPubMedGoogle Scholar
- Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, et al. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004;203:733–9. DOIPubMedGoogle Scholar
- Ironside JW, Bishop MT, Connolly K, Hegazy D, Lowrie S, Le Grice M, et al. Variant Creutzfeldt-Jakob disease: prion protein genotype analysis of positive appendix tissue samples from a retrospective prevalence study. BMJ. 2006;332:1186–8. DOIPubMedGoogle Scholar
- Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, et al. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ. 2013;347(oct15 5):f5675. DOIPubMedGoogle Scholar
- Asante EA, Linehan JM, Desbruslais M, Joiner S, Gowland I, Wood AL, et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–66. DOIPubMedGoogle Scholar
- Wadsworth JD, Asante EA, Desbruslais M, Linehan JM, Joiner S, Gowland I, et al. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306:1793–6. DOIPubMedGoogle Scholar
- Asante EA, Linehan JM, Gowland I, Joiner S, Fox K, Cooper S, et al. Dissociation of pathological and molecular phenotype of variant Creutzfeldt-Jakob disease in transgenic human prion protein 129 heterozygous mice. Proc Natl Acad Sci U S A. 2006;103:10759–64. DOIPubMedGoogle Scholar
- Bishop MT, Hart P, Aitchison L, Baybutt HN, Plinston C, Thomson V, et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006;5:393–8. DOIPubMedGoogle Scholar
- Takeuchi A, Kobayashi A, Ironside JW, Mohri S, Kitamoto T. Characterization of variant Creutzfeldt-Jakob disease prions in prion protein-humanized mice carrying distinct codon 129 genotypes. J Biol Chem. 2013;288:21659–66. DOIPubMedGoogle Scholar
- Padilla D, Béringue V, Espinosa JC, Andreoletti O, Jaumain E, Reine F, et al. Sheep and goat BSE propagate more efficiently than cattle BSE in human PrP transgenic mice. PLoS Pathog. 2011;7:e1001319 . DOIPubMedGoogle Scholar
- Espinosa JC, Herva ME, Andréoletti O, Padilla D, Lacroux C, Cassard H, et al. Transgenic mice expressing porcine prion protein resistant to classical scrapie but susceptible to sheep bovine spongiform encephalopathy and atypical scrapie. Emerg Infect Dis. 2009;15:1214–21. DOIPubMedGoogle Scholar
- Castilla J, Gutiérrez Adán A, Brun A, Pintado B, Ramírez MA, Parra B, et al. Early detection of PrPres in BSE-infected bovine PrP transgenic mice. Arch Virol. 2003;148:677–91. DOIPubMedGoogle Scholar
- Castilla J, Gutiérrez-Adán A, Brun A, Doyle D, Pintado B, Ramírez MA, et al. Subclinical bovine spongiform encephalopathy infection in transgenic mice expressing porcine prion protein. J Neurosci. 2004;24:5063–9. DOIPubMedGoogle Scholar
- Aguilar-Calvo P, Fast C, Tauscher K, Espinosa JC, Groschup MH, Nadeem M, et al. Effect of Q211 and K222 PRNP polymorphic variants in the susceptibility of goats to oral infection with goat bovine spongiform encephalopathy. J Infect Dis. 2015;212:664–72. DOIPubMedGoogle Scholar
- Foster JD, Hope J, Fraser H. Transmission of bovine spongiform encephalopathy to sheep and goats. Vet Rec. 1993;133:339–41. DOIPubMedGoogle Scholar
- Goldmann W, Martin T, Foster J, Hughes S, Smith G, Hughes K, et al. Novel polymorphisms in the caprine PrP gene: a codon 142 mutation associated with scrapie incubation period. J Gen Virol. 1996;77:2885–91. DOIPubMedGoogle Scholar
- Eloit M, Adjou K, Coulpier M, Fontaine JJ, Hamel R, Lilin T, et al. BSE agent signatures in a goat. Vet Rec. 2005;156:523–4. DOIPubMedGoogle Scholar
- Cooper JK, Ladhani K, Minor PD. Reference materials for the evaluation of pre-mortem variant Creutzfeldt-Jakob disease diagnostic assays. Vox Sang. 2007;92:302–10.PubMedGoogle Scholar
- Cassard H, Torres JM, Lacroux C, Douet JY, Benestad SL, Lantier F, et al. Evidence for zoonotic potential of ovine scrapie prions. Nat Commun. 2014;5:5821. DOIPubMedGoogle Scholar
- Notari S, Xiao X, Espinosa JC, Cohen Y, Qing L, Aguilar-Calvo P, et al. Transmission characteristics of variably protease-sensitive prionopathy. Emerg Infect Dis. 2014;20:2006–14. DOIPubMedGoogle Scholar
- Féraudet C, Morel N, Simon S, Volland H, Frobert Y, Créminon C, et al. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280:11247–58. DOIPubMedGoogle Scholar
- Andréoletti O, Berthon P, Levavasseur E, Marc D, Lantier F, Monks E, et al. Phenotyping of protein-prion (PrPsc)-accumulating cells in lymphoid and neural tissues of naturally scrapie-affected sheep by double-labeling immunohistochemistry. J Histochem Cytochem. 2002;50:1357–70. DOIPubMedGoogle Scholar
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78:301–11. DOIPubMedGoogle Scholar
- Andréoletti O, Simon S, Lacroux C, Morel N, Tabouret G, Chabert A, et al. PrPSc accumulation in myocytes from sheep incubating natural scrapie. Nat Med. 2004;10:591–3. DOIPubMedGoogle Scholar
- Wadsworth JD, Powell C, Beck JA, Joiner S, Linehan JM, Brandner S, et al. Molecular diagnosis of human prion disease. Methods Mol Biol. 2008;459:197–227. DOIPubMedGoogle Scholar
- Vidal E, Fernández-Borges N, Pintado B, Ordóñez M, Márquez M, Fondevila D, et al. Bovine spongiform encephalopathy induces misfolding of alleged prion-resistant species cellular prion protein without altering its pathobiological features. J Neurosci. 2013;33:7778–86. DOIPubMedGoogle Scholar
- de Marco MF, Linehan J, Gill ON, Clewley JP, Brandner S. Large-scale immunohistochemical examination for lymphoreticular prion protein in tonsil specimens collected in Britain. J Pathol. 2010;222:380–7. DOIPubMedGoogle Scholar
- Collinge J. Medicine. Prion strain mutation and selection. Science. 2010;328:1111–2. DOIPubMedGoogle Scholar
- Douet JY, Zafar S, Perret-Liaudet A, Lacroux C, Lugan S, Aron N, et al. Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt-Jakob disease. Emerg Infect Dis. 2014;20:114–7. DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 23, Number 9—September 2017
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Juan Maria Torres Trillo, Centro de Investigación en Sanidad Animal (CISA-INIA). 28130 Valdeolmos, Madrid, Spain
Top