Volume 25, Number 6—June 2019
Research
Phenotypic and Genomic Analyses of Burkholderia stabilis Clinical Contamination, Switzerland
Abstract
A recent hospital outbreak related to premoistened gloves used to wash patients exposed the difficulties of defining Burkholderia species in clinical settings. The outbreak strain displayed key B. stabilis phenotypes, including the inability to grow at 42°C; we used whole-genome sequencing to confirm the pathogen was B. stabilis. The outbreak strain genome comprises 3 chromosomes and a plasmid, sharing an average nucleotide identity of 98.4% with B. stabilis ATCC27515 BAA-67, but with 13% novel coding sequences. The genome lacks identifiable virulence factors and has no apparent increase in encoded antimicrobial drug resistance, few insertion sequences, and few pseudogenes, suggesting this outbreak was an opportunistic infection by an environmental strain not adapted to human pathogenicity. The diversity among outbreak isolates (22 from patients and 16 from washing gloves) is only 6 single-nucleotide polymorphisms, although the genome remains plastic, with large elements stochastically lost from outbreak isolates.
Burkholderia is a diverse genus of gram-negative bacteria, with isolates identified from a variety of environments, and ever more species being identified and classified. Whereas some Burkholderia species are associated with bioremediation potential and antimicrobial and antifungal production, others are animal and human pathogens that generally fall within the B. cepacia complex (Bcc) (1). Burkholderia bacteria have large, flexible, multi-replicon genomes, a large metabolic repertoire, various virulence factors, and inherent resistance to many antimicrobial drugs (2,3).
An outbreak of B. stabilis was identified among hospitalized patients across several cantons in Switzerland during 2015–2016 (4). The bacterium caused bloodstream infections, noninvasive infections, and wound contaminations. The source of the infection was traced to contaminated commercially available, premoistened washing gloves used for bedridden patients. After hospitals discontinued use of these gloves, the outbreak resolved.
Many instances of Bcc strain contamination of medical devices and solutions have been described (4), including an outbreak in Korea associated with a 0.5% chlorhexidine solution (5). B. stabilis also has been identified in nosocomial infections (6–8).
We conducted in-depth characterization of the B. stabilis strain from the Switzerland outbreak by using clinical methods and whole-genome sequencing (WGS). We generated a complete draft genome by combining short- and long-read genomic data and compared it to other outbreak isolates to provide a complete genomic assessment of this strain. We provide a thorough comparative genomic analysis of this outbreak strain.
Bacterial Isolate Collection
Isolates were collected from 22 patients (labeled 1–22) and 16 contaminated washing gloves (labeled A–P) across Switzerland during the outbreak (4). For comparison, we collected 14 unrelated Burkholderia spp. patient isolates in Switzerland (labeled O-1 through O-14; Appendix 1 Table 1). We obtained a control strain, B. stabilis ATCC27515 BAA-67, isolated in 1993 from sputum of a patient with cystic fibrosis in Belgium, from the American Type Culture Collection (ATCC, https://www.atcc.org).
Clinical Diagnostics
We performed routine identification using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry and biochemical species identification. For MALDI-TOF mass spectrometry, we used Biotyper MBT Smart (Bruker Corporation, https://www.bruker.com) with flexControl and MBT Compass version 4.1 software. We considered scores >2.0 high confidence identification and scores of 1.7–2.0 low confidence identification. We used VITEK 2 gram negative identification card (bioMérieux, https://www.biomerieux.com) for biochemical species identification. Phenotypic antimicrobial resistance profiles were determined using disk diffusion. We interpreted breakpoints according to Clinical and Laboratory Standards Institute (9) standards for Bcc (ceftazidime, trimethoprim/sulfamethoxazole, and meropenem) or Enterobacteriaceae (aminoglycosides, ciprofloxacin, piperacillin/tazobactam, and other β-lactams). We used XbaI to digest DNA before using previously described pulsed-field gel electrophoresis (PFGE) molecular typing principles (10). We used GelCompar (Applied Maths, http://www.applied-maths.com) to analyze PFGE results.
Cellular Fatty Acid Analysis
We prepared and derivatized cellular fatty acids from outbreak isolates 7, 13, and O, with control strain Pseudomonas aeruginosa strain ATCC27853, as previously described (11). We performed chromatography on an HP 6890 gas chromatograph (Hewlett Packard Enterprise, https://www.hpe.com) and analyzed data in SHERLOCK MIS version 6.2 (Midi Inc., http://midi-inc.com).
Genome Sequencing, Assembly, Annotation, and Mapping
We extracted DNA using EZ1 Advanced XL (QIAGEN, https://www.qiagen.com) or Wizard Genomic DNA Purification kit (Promega, https://www.promega.com) and then sequenced it on the Illumina MiSeq platform (https://www.illumina.com) following Nextera XT library creation within the Division of Clinical Microbiology, University Hospital Basel (300-bp paired-end reads) or the Unit of Genomics of the Institute of Microbiology, Lausanne University Hospital (150-bp paired-end reads). We mapped data against the genome of B. cepacia ATCC25416 (GenBank accession nos. CP007746–8) for quality control and coverage determination (Appendix 1 Table 1). We sequenced DNA from outbreak isolate 5 on a PacBio RS II platform (Pacific Biosciences, https://www.pacb.com) with 1 SMRT cell at the Functional Genomics Centre, Zurich. We submitted read data for all samples to the European Nucleotide Archive under project nos. PRJEB18658 and PRJEB19203 (data previously analyzed; 12).
We used CLC Genomics Workbench 9 (https://clc-genomics-workbench.software.informer.com/9.0) to assemble Illumina reads from outbreak isolate E (1,736 contigs; assembly length 8,816,302 bp) and from unrelated isolates. We processed PacBio reads with CLC Genomics Workbench 9 using an error correction of 30 or 50 and assembled resulting reads or used them to correct the Illumina assembly of E. We used Spades version 3.10.0 (13) to assemble the PacBio reads with Illumina reads from isolates E or 5. We manually compared assemblies in Artemis and ACT (14,15) to circularize and split chromosomes 1 and 2 according to the genome of B. stabilis BAA-67 (GenBank accession nos. CP016442–4) (16). We found chromosome 3 is a single contig that was not obviously circularizable. We used Prokka version 1.11 (17) for automated annotation and manually curated coding sequences (CDSs) by using Artemis and ACT. We submitted the genome draft to ENA under accession no. ERZ480954.
We performed mapping in CLC Genomics Workbench 9, which we also used to generate k-mer trees (18) using default parameters. For single-nucleotide polymorphism (SNP) phylogenies, we used variant calling with 5× minimum coverage, 5 minimum count, and 70% minimum frequency and created SNP trees with 5× minimum coverage, 5% minimum coverage, 0 prune distance, and multinucleotide variants.
Database and Genome Comparisons
We used multilocus sequence typing (MLST; https://cge.cbs.dtu.dk/services/MLST) to identify alleles from assemblies of isolate E and unrelated Burkholderia spp. (19) and compared these against Bcc MLST databases (https://pubmlst.org/bcc) (20) for species designation. We performed average nucleotide identity (ANI) determination using the ANI calculator (http://enve-omics.ce.gatech.edu/ani) (21), and digital DNA-DNA hybridization (dDDH) with GGDC2.1 (http://ggdc.dsmz.de/distcalc2.php) (22). We used antiSMASH version 4.0.0 (https://antismash.secondarymetabolites.org) (23) to predict gene clusters involved in antimicrobial resistance and secondary metabolite production.
Clinical and Major Fatty Acid Characterization
We analyzed outbreak and unrelated Burkholderia spp. isolates by PFGE (Appendix 2 Figure 1). PFGE patterns of outbreak isolates from patients and washing gloves formed a cluster separate from the unrelated isolates. Outbreak isolates shared 75.7% similarity by Pearson correlation analysis; previous studies used a value of 80% for outbreak grouping (24).
We conducted MALDI-TOF mass spectrometry on 12 outbreak isolates and identified B. cepacia group (n = 4), B. stabilis (n = 4), B. multivorans (n = 2), B. cenocepacia (n = 1), and B. pyrrocinia (n = 1) with scores of 1.88–2.21. Because MALDI-TOF mass spectrometry is known to misidentify Bcc species (25), we used VITEK 2 to conduct biochemical testing on isolates from 3 patients and 1 washing glove and identified either Bcc group (scores of 91%–95%) or Acinetobacter lwoffii (scores of 86%–91%).
Our outbreak isolates and control strain shared a defining characteristic of B. stabilis, the inability to grow at 42°C (26,27). Using VITEK 2, we saw 2 other key characteristics of B. stabilis in the outbreak strain, absence of β-galactosidase activity and inability to oxidize sucrose (Table 1). In contrast to other B. stabilis strains, VITEK 2 showed our strain was negative for adonitol acidification, ornithine decarboxylase, and lysine decarboxylase. Phenotypic identification of Burkholderia spp. often is a tedious process (26), with high rates of misidentification because of false negative reporting by VITEK 2 (29). Clinical standard identification on the VITEK 2 runs up to 16 hours, but Burkholderia phenotypes can take up to 7 days to develop. Our subsequent genome analysis identified genes encoding ornithine decarboxylase and lysine decarboxylase in the outbreak strain.
Cellular fatty acid profiling of 3 outbreak isolates showed profiles highly similar to the reference strains B. stabilis and B. cepacia (26,30), and expectedly distinct from control strain P. aeruginosa strain ATCC27853 (Appendix 1 Table 2; Appendix 2 Figure 2). Together, these assays identified the outbreak strain as a member of the genus Burkholderia within Bcc but did not enable a firm species-level identification.
Identification of Outbreak Isolate Clade in Bcc
We conducted WGS on 22 patient isolates and 16 isolates from washing gloves. We compared a full-length 16S rRNA gene sequence derived from the genome assembly of isolate E against the National Center for Biotechnology Information nucleotide sequence database using blastn (http://blast.ncbi.nlm.nih.gov). We identified 2 top hits, both sharing 1,520 out of 1,521 nt identities, B. pyrrocinia DSM10685, and B. stabilis BAA-67. Many other Bcc species shared >99% nucleotide identity, including B. stagnalis MSMB735WGS, B. cenocepacia AU1054, B. cenocepacia J2315, B. ambifaria AMMD, and B. lata 383. We mapped the WGS data of all outbreak isolates against a draft assembly of isolate E and found 99%–100% coverage from all isolates, but only 60%–75% coverage from unrelated isolate sequences (Appendix 1 Table 1). K-mer analysis of outbreak and unrelated Burkholderia isolates showed a separate cluster of outbreak isolates (Appendix 2 Figure 3).
Identification of New B. stabilis Strain
We used MLST to extract alleles from the genome draft to obtain a new sequence type (ST), 1095, with the following genes: atpD (380, new), gltB (456), gyrB (213), lepA (70), phaC (348, new), recA (109), trpB (173). The top matches to this MLST profile are all B. stabilis isolates sharing <4 of the 7 alleles. The matching isolates are from Canada, which had 4 matching alleles; and Czechia, Serbia, China, France, Italy, and the United Kingdom, each with 2 matching alleles, including the B. stabilis type strain ATCC27515 BAA-67.
We used ANI calculations of genomic relatedness to compare the isolate E genome draft to a comprehensive panel of sequenced Bcc strains (Table 2). B. stabilis BAA-67 is the most closely related with an ANI of 98.4%, which is above the species cutoff of 95% (31). In addition, dDDH comparing the outbreak strain to the Bcc panel showed that the maximum score of the outbreak genome is with B. stabilis BAA-67 at 84.2%, with the classic species threshold at 70%. These genomic parameters currently are the most robust for species designation (32–34) and we are confident that the outbreak strain belongs to the species B. stabilis. The high dDDH score might reflect the high genome conservation within this species, giving it its name (26).
Description of Draft of B. stabilis Strain CH16
Figure 1

Figure 1. Comparison of the genome of Burkholderia stabilis strain CH16 from Switzerland (top bar) with that of B. stabilis reference strain BAA-67 (bottom bar). Alternating orange and brown bar sections represent chromosomes...
A hybrid assembly of PacBio and Illumina data resulted in an improved, high-quality genome draft (35) of the outbreak strain, named CH16 because it occurred in Switzerland in 2016. This draft comprises 1 contig for each of the 3 chromosomes. Comparison with the genome of B. stabilis BAA-67 (16) showed that the genomes are syntenic with the exception of a rearrangement on chromosome 1 between the rRNA operons, which might be a real inversion or an assembly artifact in 1 of the genomes (Figure 1). We detected a separate contig representing a predicted plasmid sequence, whereas none was found within strain BAA-67 (J. Bugrysheva, US Centers for Disease Control and Prevention, pers. comm., 2017 Jan 10) (Table 3).
In addition to being large and multireplicon, Burkholderia genomes are characterized by the presence of multiple phages, genomic islands, and insertion sequences (IS elements) (3). The draft genome CH16 contains many insertions of single or multiple genes relative to strain BAA-67: 16 on chromosome 1; 34 on chromosome 2; and 10 on chromosome 3 (Figure 1). Appendix 1 Table 3 lists regions of difference (RDs).
The B. stabilis CH16 genome has a paucity of IS elements. We have identified only 40, including 6 families with copy numbers of 3–12 (Appendix 1 Table 4), that cause disruption of 9 CDSs (Appendix 1 Table 5). CH16 did not appear to be experiencing IS element expansion, which is associated with genome rearrangements, large-scale genomic deletions, and niche adaptation (36–38), but it has the potential for IS element expansion if it goes through a population bottleneck.
Frameshifts and premature stop codons have created 11 additional pseudogenes (Appendix 1 Table 5). The 20 pseudogenes of CH16 contrast with 142 annotated in the genome of strain BAA-67, indicating that most of the CH16 genome is required for survival in diverse environments and that this strain is not adapting to a pathogenic lifestyle.
RDs and Virulence Factors of B. stabilis Strain CH16
Using genome-wide blastn comparisons, we determined that the CH16 genome carries 973 novel CDSs relative to BAA-67 of the total 7,629 CDSs (12.7%; Appendix 1 Table 3), many of which are novel to all Burkholderia sequenced to date. Larger insertions containing >40 CDSs are putative phages or integrative and conjugative elements. Smaller insertions of <10 CDSs appear to represent deletions in the BAA-67 strain relative to their common ancestor.
Factors that might contribute to the virulence of CH16 include adhesins and hemaglutinins, including BSTAB16_1184, _5825, _5829, _5874, _6110, _6684, _6804, and _6861, of which most have homologs in other Bcc strains; and Type II and Type VI secretion systems (BSTAB16_4657–74, _5069–91, and _5583–9). The many regulators within the CH16 genome and the RDs provide additional layers of translational control necessary in a genome of this size. We saw no evidence of the known Burkholderia virulence factors cable pilus or B. cepacia epidemic strain marker (36). The toxins we identified, for example BSTAB16_5843 containing the HipA domain, are antibacterial toxins rather than virulence factors.
The many efflux pumps found in the CH16 genome might explain its ability to grow in the wash solution, including members of the following families: resistance nodulation and cell division, ATP-binding cassette, small multidrug resistance, multidrug and toxic compound extrusion, and major facilitator superfamily. Several secondary metabolite synthesis pathways are predicted: 4 on chromosome 1; 5 on chromosome 2; and 5 on chromosome 3 (Appendix 1 Table 6). Most of these are shared with the BAA-67 reference genome, encoding the ability to produce signaling molecules, siderophores, terpenes, and a bacteriocin, among others.
The plasmid comprises largely novel sequences not seen before within the Burkholderia or elsewhere. It carries genes predicted to be involved in conjugation, indicating that it might be a mobile plasmid, such as 1 recently hypothesized in B. cenocepacia (39). The rest of the plasmid largely comprises genes encoding hypothetical proteins.
Antimicrobial Drug Resistance of B. stabilis Strain CH16
We performed phenotypic antimicrobial drug susceptibility testing on a subset of outbreak isolates (Appendix 1 Table 7) and used genomic findings to interpret the results. Breakpoints are not established clinically and are not recommended to guide patient therapy (40).
All Bcc isolates are intrinsically resistant to aminoglycosides (40), which we confirmed in our isolates. Intrinsic resistance also is described against chloramphenicol and tetracycline (40) (not tested) through the presence of efflux pumps. We identified several efflux pumps within the CH16 genome (BSTAB16_5335–6, _4605–6, and _7210–1), none of which are unique to the outbreak strain. Sensitivity to trimethoprim/sulfamethoxazole was a feature of the outbreak isolates; we did not identify trimethoprim/sulfamethoxazole resistance determinants in the draft genome.
Bcc is considered to be intrinsically and clinically resistant to many β-lactams through impermeability and the presence of inducible β-lactamases (40). All Bcc isolates tested were resistant to aminopenicillins, carboxypenicillins, and first-generation cephalosporins. Phenotypic resistance to third-generation cephalosporins, ureidopenicillins, and carbapenems was more variable among Bcc. We identified several β-lactamases in the CH16 genome, representing class A (BSTAB16_4862 and _4440), class C (BSTAB16_6957), class D (BSTAB16_5918), and metallo-β-lactamases (BSTAB16_ 3974 and _5115), none of which are unique to this strain.
The outbreak isolates are sensitive to ciprofloxacin, in contrast to B. stabilis BAA-67, with sporadic resistance seen among other Bcc isolates. Resistance can be associated with efflux (40) or specific mutations in gyrA (BSTAB16_1445). The gyrA of CH16 differs from that of strain BAA-67 at I83T and A700S (numbered according to E. coli). In general, this strain does not display enhanced antimicrobial resistance compared with other clinical Bcc isolates or B. stabilis BAA-67 (2,41,42).
Of note, some of the outbreak isolates had anomalous antibiograms, which we confirmed through repeated testing (Appendix 1 Table 7). This finding might relate to colony morphology because several morphotypes were observed during clinical work on the outbreak isolates. This phenomenon is known to occur within Burkholderia (43–46), resulting from reversible colony morphotype switching (44) or stable mutations (43,46). Altered genes often are involved in exopolysaccharide production, also causing changes in biofilm production, virulence, resistance, and motility (43–46), and might result from stress (44,45).
Comparison of Outbreak Isolates
Figure 2
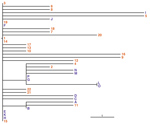
Figure 2. Phylogeny of outbreak isolates of Burkholderia stabilis strain CH16 from Switzerland based on high-quality single nucleotide polymorphisms (SNPs). This phylogeny of all sequenced outbreak isolates might represent a conservative estimate of...
PFGE and k-mer analyses showed that the outbreak isolates cluster. We investigated how related the isolates are by comparing genomes using high-quality SNPs. The SNP phylogeny (Figure 2) indicates a maximum of 6 SNPs between isolates from the outbreak source, in agreement with Abdelbary et al. (12). The previously published core genome MLST (cgMLST) phylogeny used the same genome data but indicated up to 18 alleles difference between isolates (4), though these are likely artifacts of the methodology (47).
Washing glove isolates were located throughout the phylogeny; we observed diversity within a lot number (isolates E–F and L–O), and even within single packets (isolates A, B, and L–O). Patient samples also were found throughout the phylogeny, even those originating from the same city (isolates 5, 7, 9–13 are from a first city; 6, 8, 21, and 22 are from a second city; and 14–20 are from a third city), reflecting trends seen from cgMLST data (4).
SNP locations (Appendix 1 Table 8) indicate that, of the 40 nonsynonymous SNPs, 15 are in genes predicted to encode regulators, 6 in transporters, and 3 in flagellin. We could not tell whether these are random mutations or have been subject to selective pressure, but all classes are represented in both glove and patient isolates.
In addition to the SNPs, we saw some large-scale genomic differences. By mapping read data against each individual replicon, we noted that isolates E and 20 do not carry the plasmid, which appears to be a stochastic event because the SNP phylogeny indicates that these isolates are not derived from a common ancestor. Isolates D and E have highly similar PFGE patterns (Appendix 2 Figure 1), suggesting that the plasmid does not affect the PFGE results or was lost during laboratory culture. Patient isolate 22 also shows the loss of the first 52.5 kb of chromosome 3, representing an RD, which this isolate apparently lost spontaneously during the course of the outbreak. Because of this genome plasticity, we hypothesize the CH16 genome was changing even during the course of the outbreak.
We provide a thorough and detailed description of a Burkholderia sp. outbreak resolved by WGS (4) and illustrate various associated challenges, including morphotype differences, species designation, and a large genome with associated assembly, annotation, and interpretation issues. Defining species within Bcc is notoriously difficult (26), whether phenotypically by using biochemical or MALDI-TOF mass spectrometry profiles, genomically by using 16S rRNA gene sequences, or both. WGS provides the most thorough analysis and is increasingly cost and time effective, even compared with sequencing MLST loci, interpretation of which also is complex. We saw anomalies between the phenotype of this B. stabilis outbreak strain and those described in the literature (26,27) due to shorter than optimal test incubation times in standard clinical phenotyping (29).
Several techniques can be used on WGS data to provide phylogenies. K-mer analysis (Appendix 2 Figure 3) provides an indication of clustering, but the branch lengths cannot be relied on to provide a true phylogeny and do not truly reveal relationships within clades. With this technique, a lot of genomic information regarding the coding capacity of the genome is lost. cgMLST compares nucleotide sequences of CDSs common to a group of isolates, linking isolates with the highest numbers of identical alleles. During this process some genomic data necessarily are lost, with information from accessory genes and intergenic regions disregarded. However, both methods can be performed routinely with minimal training to enable rapid visualization of outbreak clusters. Comparing reads from all outbreak isolates to an assembled draft genome to generate a SNP phylogeny includes all genomic information but requires more computation, time, and expertise.
WGS is the optimal way to determine the detailed relationships between isolates, giving insights into an outbreak and providing a basis from which to develop further typing methods. For future cases, we suggest rapid WGS, extraction of MLST alleles from assemblies for species identification as recommended by Mahenthiralingam et al. (48), and cgMLST typing for rapid outbreak identification. SNP detection can be a valuable subsequent step to determine accurate relatedness of isolates.
Bcc bacteria are known to survive in pharmaceutical and disinfectant materials (1,48,49). B. stabilis strains sharing MLST types can be isolated from the natural environment, hospitals, and patients (50), implicating the natural environment as a source of opportunistic Burkholderia and emphasizing the versatility of Bcc to survive and grow under diverse conditions. The CH16 genome displays features representative of Burkholderia in general; it is large, highly plastic, and contains many novel elements that might be involved in pathogenesis or environmental survival (36). The low number of pseudogenes and IS elements indicates that this strain has not undergone niche adaptation, and most likely is an opportunistic pathogen (36–38).
The cloud of diversity seen in the SNP phylogeny indicates that the source of the original contamination was not clonal or that several mutations occurred during the incubation of CH16 within the patient washing gloves. The loss of genomic elements, including the plasmid, from some of the isolates, also demonstrates the flexibility and the redundancy within such a large genome. Our study shows the importance of WGS in investigating and resolving this outbreak, which appears to have been caused by an environmental Bcc strain.
Dr. Seth-Smith works in clinical microbiology at University Hospital Basel, using whole-genome sequencing data to answer questions relating to emerging pathogens, outbreaks, and transmission.
Acknowledgments
We thank Clarisse Straub, Elisabeth Schultheiss, and Christine Kiessling for excellent technical assistance in performing whole-genome sequencing; Andrea Patrignani for PacBio sequencing; Doris Hohler for performing the phenotypic and antimicrobial testing; and Peter Vandamme for advice on species definition. Assemblies were performed at sciCORE (http://scicore.unibas.ch) scientific computing facility at the University of Basel.
A.E. received a research grant from the Swiss National Science Foundation (Ambizione grant no. PZ00P3_154709/1). We used the Burkholderia cepacia complex Multi Locus Sequence Typing website (http://pubmlst.org/bcc) developed by Keith Jolley (20), located at the University of Oxford and funded by the Wellcome Trust.
References
- Depoorter E, Bull MJ, Peeters C, Coenye T, Vandamme P, Mahenthiralingam E. Burkholderia: an update on taxonomy and biotechnological potential as antibiotic producers. Appl Microbiol Biotechnol. 2016;100:5215–29. DOIPubMedGoogle Scholar
- Nzula S, Vandamme P, Govan JRW. Influence of taxonomic status on the in vitro antimicrobial susceptibility of the Burkholderia cepacia complex. J Antimicrob Chemother. 2002;50:265–9. DOIPubMedGoogle Scholar
- Burns J. Antibiotic resistance of Burkholderia species. In: Coenye T, Vandamme P, editors. Burkholderia: molecular microbiology and genomics. Wymondham, England, UK: Horizon Bioscience; 2006. p. 81–91.
- Sommerstein R, Führer U, Lo Priore E, Casanova C, Meinel DM, Seth-Smith HM, et al. Burkholderia stabilis outbreak associated with contaminated commercially-available washing gloves, Switzerland, May 2015 to August 2016. Euro Surveill. 2017;22:pii:17-00213.
- Ko S, An HS, Bang JH, Park SW. An outbreak of Burkholderia cepacia complex pseudobacteremia associated with intrinsically contaminated commercial 0.5% chlorhexidine solution. Am J Infect Control. 2015;43:266–8. DOIPubMedGoogle Scholar
- Otağ F, Ersöz G, Salcioğlu M, Bal C, Schneider I, Bauernfeind A. Nosocomial bloodstream infections with Burkholderia stabilis. J Hosp Infect. 2005;59:46–52. DOIPubMedGoogle Scholar
- Heo ST, Kim SJ, Jeong YG, Bae IG, Jin JS, Lee JC. Hospital outbreak of Burkholderia stabilis bacteraemia related to contaminated chlorhexidine in haematological malignancy patients with indwelling catheters. J Hosp Infect. 2008;70:241–5. DOIPubMedGoogle Scholar
- Wang L, Wang M, Zhang J, Wu W, Lu Y, Fan Y. An outbreak of Burkholderia stabilis colonization in a nasal ward. Int J Infect Dis. 2015;33:71–4. DOIPubMedGoogle Scholar
- Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing (M100–S27). Wayne (PA); The Institute; 2017.
- Strandén A, Frei R, Widmer AF. Molecular typing of methicillin-resistant Staphylococcus aureus: can PCR replace pulsed-field gel electrophoresis? J Clin Microbiol. 2003;41:3181–6. DOIPubMedGoogle Scholar
- Osterhout GJ, Shull VH, Dick JD. Identification of clinical isolates of gram-negative nonfermentative bacteria by an automated cellular fatty acid identification system. J Clin Microbiol. 1991;29:1822–30.PubMedGoogle Scholar
- Abdelbary MMH, Senn L, Moulin E, Prod’hom G, Croxatto A, Greub G, et al. Evaluating the use of whole-genome sequencing for outbreak investigations in the lack of closely related reference genome. Infect Genet Evol. 2018;59:1–6 . DOIPubMedGoogle Scholar
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. DOIPubMedGoogle Scholar
- Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3. DOIPubMedGoogle Scholar
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream M-A, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–5. DOIPubMedGoogle Scholar
- Bugrysheva JV, Cherney B, Sue D, Conley AB, Rowe LA, Knipe KM, et al. Complete genome sequences for three chromosomes of the Burkholderia stabilis type strain (ATCC BAA-67). Genome Announc. 2016;4:e01294–16. DOIPubMedGoogle Scholar
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. DOIPubMedGoogle Scholar
- Larsen MV, Cosentino S, Lukjancenko O, Saputra D, Rasmussen S, Hasman H, et al. Benchmarking of methods for genomic taxonomy. J Clin Microbiol. 2014;52:1529–39. DOIPubMedGoogle Scholar
- Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–61. DOIPubMedGoogle Scholar
- Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. DOIPubMedGoogle Scholar
- Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91. DOIPubMedGoogle Scholar
- Auch AF, von Jan M, Klenk H-P, Göker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2010;2:117–34. DOIPubMedGoogle Scholar
- Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43(W1):
W237-43 . DOIPubMedGoogle Scholar - Chetoui H, Melin P, Struelens MJ, Delhalle E, Nigo MM, De Ryck R, et al. Comparison of biotyping, ribotyping, and pulsed-field gel electrophoresis for investigation of a common-source outbreak of Burkholderia pickettii bacteremia. J Clin Microbiol. 1997;35:1398–403.PubMedGoogle Scholar
- Fehlberg LC, Andrade LH, Assis DM, Pereira RH, Gales AC, Marques EA. Performance of MALDI-ToF MS for species identification of Burkholderia cepacia complex clinical isolates. Diagn Microbiol Infect Dis. 2013;77:126–8. DOIPubMedGoogle Scholar
- Vandamme P, Mahenthiralingam E, Holmes B, Coenye T, Hoste B, De Vos P, et al. Identification and population structure of Burkholderia stabilis sp. nov. (formerly Burkholderia cepacia genomovar IV). J Clin Microbiol. 2000;38:1042–7.PubMedGoogle Scholar
- Henry DA, Mahenthiralingam E, Vandamme P, Coenye T, Speert DP. Phenotypic methods for determining genomovar status of the Burkholderia cepacia complex. J Clin Microbiol. 2001;39:1073–8. DOIPubMedGoogle Scholar
- Vandamme P, Holmes B, Vancanneyt M, Coenye T, Hoste B, Coopman R, et al. Occurrence of multiple genomovars of Burkholderia cepacia in cystic fibrosis patients and proposal of Burkholderia multivorans sp. nov. Int J Syst Bacteriol. 1997;47:1188–200. DOIPubMedGoogle Scholar
- Zbinden A, Böttger EC, Bosshard PP, Zbinden R. Evaluation of the colorimetric VITEK 2 card for identification of gram-negative nonfermentative rods: comparison to 16S rRNA gene sequencing. J Clin Microbiol. 2007;45:2270–3. DOIPubMedGoogle Scholar
- Samuels SB, Moss CW, Weaver RE. The fatty acids of Pseudomonas multivorans (Pseudomonas cepacia) and Pseudomonas kingii. J Gen Microbiol. 1973;74:275–9. DOIPubMedGoogle Scholar
- Rodriguez-R L, Konstantinidis K. Bypassing cultivation to identify bacterial species. Microbe. 2014;9:111–8.
- Peeters C, Meier-Kolthoff JP, Verheyde B, De Brandt E, Cooper VS, Vandamme P. Phylogenomic study of Burkholderia glathei-like organisms, proposal of 13 novel Burkholderia species and emended descriptions of Burkholderia sordidicola, Burkholderia zhejiangensis, and Burkholderia grimmiae. Front Microbiol. 2016;7:877. DOIPubMedGoogle Scholar
- Meier-Kolthoff JP, Auch AF, Klenk H-P, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics. 2013;14:60. DOIPubMedGoogle Scholar
- Vandamme P, Peeters C. Time to revisit polyphasic taxonomy. Antonie van Leeuwenhoek. 2014;106:57–65. DOIPubMedGoogle Scholar
- Chain PS, Grafham DV, Fulton RS, Fitzgerald MG, Hostetler J, Muzny D, et al.; Genomic Standards Consortium Human Microbiome Project Jumpstart Consortium. Genomics. Genome project standards in a new era of sequencing. Science. 2009;326:236–7. DOIPubMedGoogle Scholar
- Holden MT, Seth-Smith HM, Crossman LC, Sebaihia M, Bentley SD, Cerdeño-Tárraga AM, et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J Bacteriol. 2009;191:261–77. DOIPubMedGoogle Scholar
- Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, et al. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet. 2003;35:32–40 . DOIPubMedGoogle Scholar
- Stinear TP, Seemann T, Harrison PF, Jenkin GA, Davies JK, Johnson PD, et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008;18:729–41. DOIPubMedGoogle Scholar
- Fernández-González E, Bakioui S, Gomes MC, O’Callaghan D, Vergunst AC, Sangari FJ, et al. A Functional oriT in the Ptw Plasmid of Burkholderia cenocepacia Can Be Recognized by the R388 Relaxase TrwC. Front Mol Biosci. 2016;3:16. DOIPubMedGoogle Scholar
- European Committee on Antimicrobial Susceptibility Testing. Antimicrobial susceptibility of Burkholderia cepacia complex (BCC). Stockholm: EUCAST; 2013 [cited 01 Dec 2017]. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/General_documents/BCC_susceptibility_testing_130719.pdf
- Peeters E, Nelis HJ, Coenye T. In vitro activity of ceftazidime, ciprofloxacin, meropenem, minocycline, tobramycin and trimethoprim/sulfamethoxazole against planktonic and sessile Burkholderia cepacia complex bacteria. J Antimicrob Chemother. 2009;64:801–9. DOIPubMedGoogle Scholar
- Tseng S-P, Tsai W-C, Liang C-Y, Lin Y-S, Huang J-W, Chang C-Y, et al. The contribution of antibiotic resistance mechanisms in clinical Burkholderia cepacia complex isolates: an emphasis on efflux pump activity. PLoS One. 2014;9:
e104986 . DOIPubMedGoogle Scholar - Silva IN, Ferreira AS, Becker JD, Zlosnik JE, Speert DP, He J, et al. Mucoid morphotype variation of Burkholderia multivorans during chronic cystic fibrosis lung infection is correlated with changes in metabolism, motility, biofilm formation and virulence. Microbiology. 2011;157:3124–37. DOIPubMedGoogle Scholar
- Chantratita N, Wuthiekanun V, Boonbumrung K, Tiyawisutsri R, Vesaratchavest M, Limmathurotsakul D, et al. Biological relevance of colony morphology and phenotypic switching by Burkholderia pseudomallei. J Bacteriol. 2007;189:807–17. DOIPubMedGoogle Scholar
- Zlosnik JE, Hird TJ, Fraenkel MC, Moreira LM, Henry DA, Speert DP. Differential mucoid exopolysaccharide production by members of the Burkholderia cepacia complex. J Clin Microbiol. 2008;46:1470–3. DOIPubMedGoogle Scholar
- Bernier SP, Nguyen DT, Sokol PA. A LysR-type transcriptional regulator in Burkholderia cenocepacia influences colony morphology and virulence. Infect Immun. 2008;76:38–47. DOIPubMedGoogle Scholar
- Leekitcharoenphon P, Nielsen EM, Kaas RS, Lund O, Aarestrup FM. Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS One. 2014;9:
e87991 . DOIPubMedGoogle Scholar - Mahenthiralingam E, Baldwin A, Dowson CG. Burkholderia cepacia complex bacteria: opportunistic pathogens with important natural biology. J Appl Microbiol. 2008;104:1539–51. DOIPubMedGoogle Scholar
- Haim MS, Mollerach M, Van Domselaar G, Teves SA, Degrossi J, Cardona ST. Draft Genome Sequences of Burkholderia contaminans FFI-28, a strain isolated from a contaminated pharmaceutical solution. Genome Announc. 2016;4:e01177–16. DOIPubMedGoogle Scholar
- Baldwin A, Mahenthiralingam E, Drevinek P, Vandamme P, Govan JR, Waine DJ, et al. Environmental Burkholderia cepacia complex isolates in human infections. Emerg Infect Dis. 2007;13:458–61. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: April 23, 2019
1Current affiliation: Genetics and Genomics at Roche, Basel, Switzerland.
2Current affiliation: RWTH Aachen University Hospital, Aachen, Germany.
3Current affiliation: University of Bern, Bern, Switzerland
Table of Contents – Volume 25, Number 6—June 2019
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Helena M.B. Seth-Smith or Adrian Egli, Universitätsspital Basel, Division of Clinical Bacteriology and Mycology, Petersgraben 4, Basel 4031, Switzerland; or
Top