Volume 26, Number 10—October 2020
Dispatch
Limitations of Ribotyping as Genotyping Method for Corynebacterium ulcerans
Abstract
We conducted molecular typing of a Corynebacterium ulcerans isolate from a woman who died in Japan in 2016. Genomic DNA modification might have affected the isolate’s ribotyping profile. Multilocus sequence typing results (sequence type 337) were more accurate. Whole-genome sequencing had greater ability to discriminate lineages at high resolution.
Corynebacterium ulcerans is a zoonotic pathogen that causes an illness categorized in World Health Organization documents as diphtheria (1). Genotyping methods such as ribotyping, multilocus sequence typing (MLST), and whole-genome sequencing are used to classify isolates. During the 1990s and early 2000s, the standard molecular typing method of Corynebacterium diphtheriae was conventional ribotyping (2,3). Ribotyping is also used to classify C. ulcerans (4) and compare isolates (5–9). Today, the standard method is MLST because of its objectivity and reproducibility (8,10). We sequenced 3 isolates of C. ulcerans from patients in Japan to analyze the accuracy of conventional ribotyping, MLST, and whole-genome sequencing.
In 2016, a 66-year-old woman in Fukuoka, Japan, died of a diphtheria-like disease. Otsuji et al. isolated toxigenic C. ulcerans from the patient’s tracheal pseudomembrane and blood (6). We analyzed the isolate (FH2016-1) from the pseudomembrane alongside the first (11) and second (5) C. ulcerans isolates taken from patients in Japan; the first isolate (0102) was taken in 2001 and the second isolate (0211) in 2002.
Figure 1
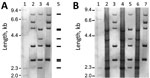
Strains 0102 and 0211 (named for the initial isolates taken in 2001 and 2002) are the 2 major ribotypes of C. ulcerans in Japan. Our conventional ribotyping of the isolates found the pattern obtained from FH2016-1 was indistinguishable from that of 0102, indicating that FH2016-1 belongs to strain 0102 (Figure 1, panel A).
We also whole-genome sequenced strains FH2016-1 and 0211 using the NextSeq500 Illumina (for strain FH2016-1 [Illumina, https://www.illumina.com]), Illumina GAII (for strain 0211 [Illumina]), ABI 3730xl (Thermo Fisher, https://www.thermofisher.com), and PacBio Sequel (Pacific Biosciences of California, Inc., https://www.pacb.com) sequencers, followed by de novo assembly. We deposited complete sequences and assembly methods in GenBank under accession nos. AP019663 (strain FH2016-1) and AP019662 (strain 0211). Using these sequences and the previously published genome sequence (12) of strain 0102 (GenBank accession no. AP012284), we conducted in silico ribotyping of BstEII-digested fragments that hybridized with OligoMix5 probes, producing a predicted pattern for each sequence (13). The predicted patterns of all 3 strains matched the conventional ribotype pattern of strain 0211. However, the conventional ribotyping patterns of strains FH2016-1 and 0102 did not match the in silico–predicted ribotype pattern (Figure 1, panel A).
The discrepancy between the conventional and in silico–predicted patterns is caused by impaired restriction digestion at specific BstEII sites. In these strains, the conventional (modified) ribotype pattern differed from the in silico–predicted (unmodified) ribotype pattern by a shift of 4 fragments (Appendix Figure 1, panel A). For example, in silico typing predicted that 3 BstEII sites would be digested at nt 770,000 of strain FH2016-1. PacBio modification analysis revealed that 1 of these sites might have been modified (Appendix Figure 1, panel B). BstEII is sensitive to methylation and other types of DNA modification (14). Thus, the difference in restriction fragment patterns was closely related to the nucleotide modifications within BstEII recognition sites (Appendix Figure 1, panel B). Other BstEII sites also might have been modified, resulting in the 4-fragment shift. Accordingly, we did not observe this shift in ribotypes of unmodified DNA substrate prepared by whole-genome amplification of the 3 strains (15) (Figure 1, panel B). The patterns of unmodified DNA matched the pattern of strain 0211 (Figure 1, panel B) and the in silico–predicted pattern (Figure 1, panel A). The >6.1-kb bands seen in “native” lanes were not visible in whole-genome amplification lanes, potentially because of the failure of whole-genome amplification to generate large fragments. These results indicate that ribotyping patterns might be substantially affected by DNA modification.
Figure 2
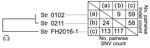
Figure 2. Genetic similarity among 3 selected strains of Corynebacterium ulcerans, Japan, 2001–2016. Strain 0102 is represented by (a), strain 0211 by (b), and strain FH2016–1 by (c). Numbers of...
The sequences of strains FH2016-1, 0102, and 0211 were highly homologous. For example, they shared complete sequence identity (data not shown) for a structural gene (locus tag CULCFH20161_03390) encoding a DNA methylase. However, we observed small differences in their genomes (Table; Figure 2; Appendix Table 1). We expected factors contributing to genomic DNA modification to be common between strains FH2016-1 and 0102, but not 0211. Scanning the genomes of the 3 strains for such factors resulted in 15 candidate open reading frames (ORFs) (Table). None of these ORFs contained motifs related to DNA methylation; however, these ORFs might still contribute to DNA modification of other gene products. The nature of the modification(s) remains unknown.
Conventional ribotyping (Figure 1, panel A) showed that strains FH2016-1 and 0102 were closely related. However, comparison of 30 genome sequences of strains from around the world (Appendix Table 2, Figure 2) revealed that all 3 strains from Japan belong to a single phylogenetic cluster and sequence type (ST) 337. Whether the 3 isolates represent the entire population of C. ulcerans in Japan is unclear. However, more than half the isolates we have analyzed (»20) are ST337 (M. Iwaki and A. Yamamoto, unpub. data), suggesting a small amount of genetic diversity among the C. ulcerans population in Japan.
Close-up view of the phylogenetic tree showed that these strains from Japan divided into 2 different lineages. At most, 117 single nucleotide variations and 59 insertions/deletions existed between any 2 strains (Figure 2). Although this result indicated low variability among the 3 strains, it also showed that strain FH2016-1 was genetically distinct from 0102 and 0211 (Figure 2). Thus, the genome sequence analysis indicated that conventional ribotyping did not reflect lineage accurately and resulted in a misleading classification of these specimens. In contrast, MLST, which is now the preferred method of molecular typing (8,10), provided more accurate results. We queried the genomic sequences of the 3 strains on the PubMLST website (https://pubmlst.org) and analyzed them at 7 alleles (atpA, dnaE, dnaK, fusA, leuA, odhA, and rpoB). The same sequence type (ST337) was assigned to all 3 strains, reflecting the low genetic variability among these strains.
Our study of 3 strains of C. ulcerans showed that conventional ribotyping is less accurate than other methods of phylogenetic analysis. In comparison, MLST is less erroneous, and whole-genome sequencing produces results with greater resolution than those of conventional ribotyping. MLST produced results with lower resolution than whole-genome sequencing while maintaining a high level of accuracy. MLST and whole-genome sequencing improve the accuracy and efficiency of phylogenetic analysis of C. ulcerans.
Dr. Sekizuka is chief at Laboratory of Bacterial Genomics, Pathogen Genomics Center, National Institute of Infectious Diseases (Shinjuku-ku, Tokyo). His primary research interests include genomic molecular epidemiology with genomics and bioinformatics, and pathogen identification with metagenomics.
Acknowledgments
We are grateful to Kaoru Umeda for her comments on the manuscript.
This research was supported by the Japan Agency for Medical Research and Development under grant nos. JP18fk0108017, JP19fk0108097, and JP19fk0108103.
References
- World Health Organization. Diphtheria. In: Vaccine preventable diseases surveillance standards. Geneva: The Organization; 2018.
- De Zoysa A, Hawkey P, Charlett A, Efstratiou A. Comparison of four molecular typing methods for characterization of Corynebacterium diphtheriae and determination of transcontinental spread of C. diphtheriae based on BstEII rRNA gene profiles. J Clin Microbiol. 2008;46:3626–35. DOIPubMedGoogle Scholar
- Grimont PAD, Grimont F, Efstratiou A, De Zoysa A, Mazurova I, Ruckly C, et al.; European Laboratory Working Group on Diphtheria. International nomenclature for Corynebacterium diphtheriae ribotypes. Res Microbiol. 2004;155:162–6. DOIPubMedGoogle Scholar
- De Zoysa A, Hawkey PM, Engler K, George R, Mann G, Reilly W, et al. Characterization of toxigenic Corynebacterium ulcerans strains isolated from humans and domestic cats in the United Kingdom. J Clin Microbiol. 2005;43:4377–81. DOIPubMedGoogle Scholar
- Komiya T, Seto Y, De Zoysa A, Iwaki M, Hatanaka A, Tsunoda A, et al. Two Japanese Corynebacterium ulcerans isolates from the same hospital: ribotype, toxigenicity and serum antitoxin titre. J Med Microbiol. 2010;59:1497–504. DOIPubMedGoogle Scholar
- Otsuji K, Fukuda K, Endo T, Shimizu S, Harayama N, Ogawa M, et al. The first fatal case of Corynebacterium ulcerans infection in Japan. JMM Case Rep. 2017;4:
e005106 . DOIPubMedGoogle Scholar - Yasuda I, Matsuyama H, Ishifuji T, Yamashita Y, Takaki M, Morimoto K, et al. Severe pneumonia caused by toxigenic Corynebacterium ulcerans infection, Japan. Emerg Infect Dis. 2018;24:588–91. DOIPubMedGoogle Scholar
- Katsukawa C, Komiya T, Umeda K, Goto M, Yanai T, Takahashi M, et al. Toxigenic Corynebacterium ulcerans isolated from a hunting dog and its diphtheria toxin antibody titer. Microbiol Immunol. 2016;60:177–86. DOIPubMedGoogle Scholar
- Katsukawa C, Umeda K, Inamori I, Kosono Y, Tanigawa T, Komiya T, et al. Toxigenic Corynebacterium ulcerans isolated from a wild bird (ural owl) and its feed (shrew-moles): comparison of molecular types with human isolates. BMC Res Notes. 2016;9:181. DOIPubMedGoogle Scholar
- König C, Meinel DM, Margos G, Konrad R, Sing A. Multilocus sequence typing of Corynebacterium ulcerans provides evidence for zoonotic transmission and for increased prevalence of certain sequence types among toxigenic strains. J Clin Microbiol. 2014;52:4318–24. DOIPubMedGoogle Scholar
- Hatanaka A, Tsunoda A, Okamoto M, Ooe K, Nakamura A, Miyakoshi M, et al. Corynebacterium ulcerans Diphtheria in Japan. Emerg Infect Dis. 2003;9:752–3. DOIPubMedGoogle Scholar
- Sekizuka T, Yamamoto A, Komiya T, Kenri T, Takeuchi F, Shibayama K, et al. Corynebacterium ulcerans 0102 carries the gene encoding diphtheria toxin on a prophage different from the C. diphtheriae NCTC 13129 prophage. BMC Microbiol. 2012;12:72. DOIPubMedGoogle Scholar
- Regnault B, Grimont F, Grimont PAD. Universal ribotyping method using a chemically labelled oligonucleotide probe mixture. Res Microbiol. 1997;148:649–59. DOIPubMedGoogle Scholar
- Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015;43(D1):D298–9. DOIPubMedGoogle Scholar
- Nelson JR. Random-primed, Phi29 DNA polymerase-based whole genome amplification. Curr Protoc Mol Biol. 2014;105:15.13.1.
Figures
Table
Cite This ArticleOriginal Publication Date: September 03, 2020
1Current affiliation: Osaka Prefecture University, Osaka, Japan.
Table of Contents – Volume 26, Number 10—October 2020
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Masaaki Iwaki, Department of Bacteriology II and Management Department of Biosafety and Laboratory Animal, National Institute of Infectious Diseases, 4-7-1 Gakuen, Musashimurayama-shi, Tokyo 208-0011, Japan
Top