Volume 27, Number 12—December 2021
Research Letter
Real-Time Projections of SARS-CoV-2 B.1.1.7 Variant in a University Setting, Texas, USA
Abstract
We used the incidence of spike gene target failures identified during PCR testing to provide an early projection of the prevalence of severe acute respiratory syndrome coronavirus 2 variant B.1.1.7 in a university setting in Texas, USA, before sequencing results were available. Findings from a more recent evaluation validated those early projections.
Identification of the highly transmissible novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variant B.1.1.7 (Alpha variant) in the United Kingdom raised concerns for renewed pandemic surges worldwide (1,2). B.1.1.7 likely arrived in the United States by October 2020 (1); it was first detected in December 2020 and declared the dominant strain in April 2021, as projected in January 2021 (3). However, the regional prevalence of B.1.1.7 was largely unknown in early 2021 because of limited molecular surveillance for SARS-CoV-2 (4). To provide local situational awareness at that pivotal moment in the coronavirus disease (COVID-19) pandemic, we estimated the prevalence of B.1.1.7 on the basis of 17,003 student SARS-CoV-2 PCR test results reported through the Proactive Community Testing program at the University of Texas (UT; Austin, Texas, USA), a large public university located in a metropolitan area with a population >2 million, during January 16–February 12, 2021 (K.E. Johnson et al., unpub. data, https://doi.org/10.1101/2021.03.05.21252541). Those early estimates were subsequently validated by using PCR data through April 9, 2021.
Mutations in the B.1.1.7 spike protein result in a failure to detect the spike gene probe in standard SARS-CoV-2 quantitative reverse transcription PCR (qRT-PCR). In estimating the prevalence of B.1.1.7 from local quantitative PCR data, we initially assumed US estimates for the proportion of spike gene target failures (SGTF) attributable to B.1.1.7 (4) and, in our retrospective analysis, update that proportion on the basis of local sequencing data. We used a Bayesian model to estimate the local growth rate of B.1.1.7 among all SARS-CoV-2 infections and applied a compartmental susceptible-exposed-infected-recovered model of SARS-CoV-2 transmission to project the effect of B.1.1.7 on future COVID-19 prevalence.
We previously estimated that the relative frequency of B.1.1.7 among positive SARS-CoV-2 samples was growing logistically at a daily rate of 0.077 (95% CI 0.017–0.140), corresponding to an early doubling time of 9.0 days (95% CI 5.0–41.0 days) (K.E. Johnson et al., unpub. data, https://doi.org/10.1101/2021.03.05.21252541). At the time, we projected that B.1.1.7 would comprise most cases at UT by March 5 (95% predictive interval [PI] February 20–March 28) (Figure, panel A).
Figure
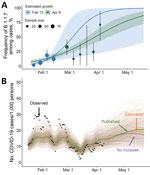
Figure. Estimated frequency of the B.1.1.7 variant among COVID-19 cases at the University of Texas and its projected impact on COVID-19 prevalence, Texas, USA, January 16–May 23, 2021. A) On the...
Subsequent estimates of B.1.1.7 prevalence based on quantitative PCR data from February 20 through April 9 fell within 95% PIs of the early projections (Figure, panel A) but suggested a lower daily growth rate of 0.037 (95 CI 0.026–0.048) and a corresponding doubling time of 18.7 days (95% CI 14.3–26.7 days). As of April 9, we estimated that B.1.1.7 comprised 61.2% (95% CI 48.5%–72.6%) of SARS-CoV-2 infections, consistent with our initial projections that B.1.1.7 would become the dominant variant by March 28 (95% CI March 20–April 10) and that B.1.1.7 is 24% (95% CI 17%–32%) more transmissible than the wild-type virus.
Based on those local estimates, scenario-based projections suggested that B.1.1.7 might cause 6.2% (95% PI 3.7%–8.4%) more cumulative infections during April 9–May 23, 2021, than if it were not more transmissible than the wild-type virus (Figure, panel B). When we assume a higher published estimate for the relative transmissibility of B.1.1.7 of 59% (95% CI 56%–63%) (2), we projected that B.1.1.7 would increase overall incidence by 14.3% (95% CI 10.8%–18.0%) during this period (Figure, panel B; Appendix Figure 5). We provide projections as total infections, rather than hospitalizations or deaths, because the primary concerns of the university at the time of this analysis were anticipating increased demand for isolation facilities, testing, and contact tracing. In either scenario, if behavior stays constant for the remainder of the semester, then we would not expect B.1.1.7 to drive a major surge in infections in the university community during this period (Figure, panel B). The relatively small effect derives from 2 factors that constrained future growth of B.1.1.7. We estimated that, by April 9, 47% (95% CI 39%–57%) of the student community was immunized by prior infection (either viral variant providing complete immunity) and that B.1.1.7 already comprised most (61.2%) new cases. This result hinges on the assumption that previous infection from either viral variant confers immunity to both variants and therefore would not apply to any type able to evade vaccine- or infection-acquired immunity. Our projections, which do not consider future behavioral change or reflect the full range of uncertainty, were not intended as forecasts but rather as plausible guideposts to help the university anticipate the severity of B.1.1.7.
UT surveillance testing indicates that B.1.1.7 rapidly became the dominant variant during the spring 2021 semester. Our methodology enabled rapid detection of B.1.1.7 emergence from widely available quantitative PCR data when sequence confirmation was not available or delayed, while quantifying uncertainty in the variant growth rate and fraction of SGTF samples that were positive for B.1.1.7. During January 16–March 5, UT confirmed 22 of 23 sequenced SGTF SARS-CoV-2 specimens as the B.1.1.7 variant, corroborating our reliance on SGTF data (Appendix).
Our findings reinforce the urgent need for expanded molecular surveillance capacity. In the absence of widespread and rapid sequencing efforts, quantitative PCR data from large-scale testing efforts have provided sentinel warning of B.1.1.7 emergence in cities throughout the United States.
Dr. Johnson is a postdoctoral researcher in the Department of Integrative Biology at the University of Texas at Austin. She uses mathematical models to aid in decision-making for control of infectious diseases. Dr. Woody is a postdoctoral researcher in the Department of Integrative Biology at the University of Texas at Austin. He develops mathematical and statistical models to elucidate transmission dynamics, surveillance, and control of infectious diseases.
Acknowledgments
We thank Andreas Matouschek for his insightful comments on the manuscript. We also thank the Proactive Community Testing team at the University of Texas at Austin.
We received financial support from the National Institutes of Health (grant no. R01 AI151176) and the Centers for Disease Control and Prevention (grant no. U01IP001136).
References
- Du Z, Wang L, Yang B, Ali ST, Tsang TK, Shan S, et al. Risk for international importations of variant SARS-CoV-2 originating in the United Kingdom. Emerg Infect Dis. 2021;27:1527–9. DOIPubMedGoogle Scholar
- Davies NG, Abbott S, Barnard RC, Jarvis CI, Kucharski AJ, Munday JD, et al.; CMMID COVID-19 Working Group; COVID-19 Genomics UK (COG-UK) Consortium. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372:
eabg3055 . DOIPubMedGoogle Scholar - Galloway SE, Paul P, MacCannell DR, Johansson MA, Brooks JT, MacNeil A, et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage - United States, December 29, 2020-January 12, 2021. MMWR Morb Mortal Wkly Rep. 2021;70:95–9. DOIPubMedGoogle Scholar
- Washington NL, Gangavarapu K, Zeller M, Bolze A, Cirulli ET, Schiabor Barrett KM, et al. Emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell. 2021;184:2587–2594.e7. DOIPubMedGoogle Scholar
- University of Texas at Austin. University of Texas Proactive Community Testing Program for COVID-19 [cited 2021 Feb 9]. https://healthyhorns.utexas.edu/coronavirus_proactive_testing.html
- Cori A, Ferguson NM, Fraser C, Cauchemez S. A new framework and software to estimate time-varying reproduction numbers during epidemics. Am J Epidemiol. 2013;178:1505–12. DOIPubMedGoogle Scholar
- Volz E, Mishra S, Chand M, Barrett JC, Johnson R, Geidelberg L, et al.; COVID-19 Genomics UK (COG-UK) consortium. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature. 2021;593:266–9. DOIPubMedGoogle Scholar
Figure
Cite This ArticleOriginal Publication Date: October 27, 2021
1These authors contributed equally to this article.
Table of Contents – Volume 27, Number 12—December 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Lauren Ancel Meyers, The University of Texas at Austin, Department of Integrative Biology, 1 University Station C0930, Austin, TX 78712, USA
Top