Volume 27, Number 12—December 2021
Dispatch
SARS-CoV-2 Variants, South Sudan, January–March 2021
Abstract
As the coronavirus pandemic continues, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) sequence data are required to inform vaccine efforts. We provide SARS-CoV-2 sequence data from South Sudan and document the dominance of SARS-CoV-2 lineage B.1.525 (Eta variant) during the country's second wave of infection.
As of August 2021, coronavirus disease (COVID-19) had caused >199 million cases and >4.2 million deaths worldwide (1). Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes COVID-19, is being sequenced to document virus evolution and to inform vaccine efforts. In South Sudan, the COVID-19 index case was confirmed on April 4, 2020 (2); it was followed by 2 infection waves, in May–July 2020 and in February–March 2021 (Appendix Figure 1). As of August 3, 2021, a total of 11,063 cases and 119 deaths (1) had been reported in South Sudan. An earlier study from South Sudan reported that, after the second wave, 28% of the population showed serologic evidence of infection (3).
Chronic underdevelopment caused by prolonged conflicts has left South Sudan with a weak health system and population displacement. Large populations live in camps that may promote rapid spread and amplification of SARS-CoV-2, and poor socioeconomic conditions limit community-based COVID-19 prevention efforts. Monitoring the circulating viral genomic lineages in South Sudan is crucial, especially as vaccination is implemented and novel virus variants appear globally.
Figure 1
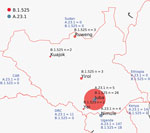
Figure 1. Locations of severe acute respiratory syndrome coronavirus 2 infection case-patients from whom genomes were isolated, South Sudan. Red circles indicate viruses of lineage B.1.525; dark gray circles indicate lineage A.23.1....
As part of South Sudan COVID-19 surveillance, samples were collected from community surveillance, point-of-entry screening, and sentinel site surveillance and tested for SARS-CoV-2 by real-time reverse transcription PCR (RT-PCR) at the National Public Health Laboratory (Juba, South Sudan) (4). During the second COVID-19 wave in February–March 2021 (Appendix Figure 1), we tested 56,014 samples for SARS-CoV-2; 6,645 samples tested positive (12% positivity). We selected a set of 70 (1%) of these positive samples for genomic sequencing with these inclusion criteria: diagnostic RT-PCR cycle threshold (Ct) values <31, from multiple locations (Figure 1), from new arrivals, from death cases, and from sites showing community transmission. We extracted nucleic acid from swab material and generated SARS-CoV-2 genome as previously described (5). A total of 45 complete genomes generated from samples collected in January–March 2021 showed a prevalence of 2 lineages: B.1.525 (Eta) and A.23.1 (Figure 1). The A.23.1 lineage, which was observed in October 2020 in Uganda (6) and has now spread globally to 26 countries, was one we observed in Juba and Nimule in early January 2021. The A.23.1 case-patients in Nimule, a South Sudan town on the border with Uganda (Figure 1), were travelers returning from Uganda. In Juba, the earliest-reported A23.1 case was in a traveler returning to South Sudan from Uganda. We detected A23.1 for only a short period; from the end of January to the end of March, we detected only B.1.525 genomes (Appendix Table). The B.1.525 lineage, reported earliest in the United Kingdom and Nigeria, has spread to 44 countries and is considered a variant of interest (7).
Figure 2

Figure 2. Maximum-likelihood phylogenetic tree of severe acute respiratory syndrome coronavirus 2 viruses from South Sudan (red dots) and reference sequences. A) Lineage A.23.1. All sequences from South Sudan were combined with...
Phylogenetic analyses of the South Sudan genomes combined with the available global A.23.1 or B.1.525 genomes were performed to gain insight into the virus movement. The maximum-likelihood trees of both A.23.1 and B.1.525 genome sequences (Figure 2) suggested multiple importations of the strains into South Sudan; South Sudan strains belonged to several sublineages, rather than a single sublineage.
Both A.23.1 and B.1.525 lineages encoded changes in their spike protein (Appendix Figure 2) as well as other parts of the genome and substitutions or deletions in the nonstructural protein 6, open reading frame 3a and 8, and nucleocapsid genes (data not shown), which might be associated with higher transmission or immune evasion. Especially relevant, the A.23.1 genomes encoded spike P681R, which is adjacent to the small (S) 1/S2 furin cleavage site and is also present in the variant of concern B.1.617.2 (Delta) lineage, which is spreading in India and globally and may increase S1/S2 cleavage (10,11; B. Lubinski et al., unpub. data. http://biorxiv.org/lookup/doi/10.1101/2021.06.30.450632). A related P681H substitution is present in variants of concern B.1.1.7 (Alpha) and P.1 (Beta). The South Sudan B.1.525 genomes encoded a deletion in the N-terminal domain (NTD) at spike positions 69 and 79, which is also present in B.1.1.7 and many other global variants, and a deletion in the spike NTD in positions 141–146, which may help in evasion of host immune responses. The spike D614G substitution may alter the spike protein conformation; the Q677H substitution is near the furin cleavage site and may alter spike processing.
We describe the patterns of SARS-CoV-2 virus genomics in South Sudan in the second wave of infections during February–March 2021, showing circulation of B.1.525 (Eta) as well as the variant A.23.1. South Sudan faced high transmission of SARS-CoV-2 during this reporting period; our data suggest that the B.1.525 lineage spread widely and progressively increased in frequency in the country during the period. Data from Uganda and Rwanda retrieved from GISAID (https://www.gisaid.org) also showed the appearance of B.1.525 at this time.
A limitation of our study is that sample numbers are low and were limited by the challenges of procurement, shipment, and testing in a harsh and resource-poor environment. Careful sample selection was performed to provide an unbiased description of the epidemic; however, not all positive samples yielded genome sequences. This lack of data could introduce bias in the reported genomes. Nonetheless, the study accurately describes SARS-CoV-2 lineages during the second wave of epidemic in South Sudan.
Substantial land-based traffic with neighboring countries makes it imperative to document the viruses circulating in this region. Careful monitoring of locally circulating viruses as vaccination becomes widespread is essential for interpreting vaccine function and for informing the healthcare systems whether the current vaccines are still a good match for the circulating viruses. We recommend continued genomic surveillance in South Sudan to help with public health responses, especially as new waves of infections come to the country and continent.
Dr. Bugembe is a scientist at the Medical Research Council/Uganda Virus Research Institute and London School of Hygiene and Tropical Medicine Research Unit in Entebbe, Uganda. His research interests are virus evolution and the computational analysis of viral genetic data to predict infectious disease trends.
Acknowledgment
D.L.B., M.V.T.P., and M.C. were funded by the Medical Research Council/United Kingdom Research and Innovation (grant no. NC_PC_19060) and Wellcome Trust (grant no. 220977/Z/20/Z).
References
- Center for Systems Science and Engineering (CSSE) at Johns Hopkins University. COVID-19 dashboard. 2020 [cited 2021 Jan 24]. https://www.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6
- World Health Organization. South Sudan confirms first case of COVID-19. 2020 [cited 2021 Sep 3]. https://www.afro.who.int/news/south-sudan-confirms-first-case-covid-19
- Wiens KE, Mawien PN, Rumunu J, Slater D, Jones FK, Moheed S, et al. Seroprevalence of severe acute respiratory syndrome coronavirus 2 IgG in Juba, South Sudan, 2020. Emerg Infect Dis. 2021;27:1598–606. DOIPubMedGoogle Scholar
- World Health Organization. South Sudan joint external evaluation report 2018. 2018 [cited 2021 Sep 3]. https://www.afro.who.int/publications/south-sudan-joint-external-evaluation-report2018
- Cotten M, Bugembe DL, Kaleebu P, Phan , MVT. Alternate primers for whole-genome SARS-CoV-2 sequencing. Virus Evol. 2021; 27:veab006.
- Bugembe DL, Phan MVT, Ssewanyana I, Semanda P, Nansumba H, Dhaala B, et al. Emergence and spread of a SARS-CoV-2 lineage A variant (A.23.1) with altered spike protein in Uganda. Nat Microbiol. 2021;6:1094–101. DOIPubMedGoogle Scholar
- World Health Organization. Tracking SARS-CoV-2 variants. 2021 [cited 2021 Sep 3]. https://www.who.int/en/activities/tracking-SARS-CoV-2-variants
- Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics. 2019;35:4453–5. DOIPubMedGoogle Scholar
- Darriba D, Posada D, Kozlov AM, Stamatakis A, Morel B, Flouri T. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol Biol Evol. 2020;37:291–4. DOIPubMedGoogle Scholar
- Johnson BA, Xie X, Bailey AL, Kalveram B, Lokugamage KG, Muruato A, et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature. 2021;591:293–9. DOIPubMedGoogle Scholar
- Hoffmann M, Kleine-Weber H, Pöhlmann S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells.
Figures
Cite This ArticleOriginal Publication Date: October 27, 2021
Table of Contents – Volume 27, Number 12—December 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Matthew Cotten, MRC/UVRI & LSHTM Uganda Research Unit, PO Box 49, Plot 51-59 Nakiwogo Rd, Entebbe, Uganda
Top