Volume 28, Number 7—July 2022
Dispatch
Isolation and Characterization of Novel Reassortant Influenza A(H10N7) Virus in a Harbor Seal, British Columbia, Canada
Abstract
We isolated a novel reassortant influenza A(H10N7) virus from a harbor seal in British Columbia, Canada, that died from bronchointerstitial pneumonia. The virus had unique genome constellations involving lineages from North America and Eurasia and polymerase basic 2 segment D701N mutation, associated with adaptation to mammals.
Influenza A viruses (IAVs) are one of the most consequential pathogens of birds and terrestrial mammals. IAVs (family Orthomyxoviridae) are enveloped, segmented, negative-sense, single-stranded RNA viruses, subtyped based on the surface glycoproteins hemagglutinin (H) and neuraminidase (N) embedded in the virus envelope. With the exceptions of H17N10 and H18N11 IAVs in bats, all other existing combinations of H (n = 16) and N (n = 9) subtypes are found in wild aquatic and shore birds, their natural reservoir hosts (1).
Phylogenetically, IAVs can be separated into 2 genetically distinct lineages, Eurasia (EA) and North America (NA), because of the fidelity of migratory waterfowl to distinct flyways (2). However, in areas where migratory flyways overlap, there are, although rare, IAVs with gene segments from both NA and EA lineages. Surveillance studies in Alaska (United States) and Newfoundland (Canada) documented a preponderance of IAVs with whole-genome segments from EA lineage viruses and only rarely, reassortant IAVs with EA and NA gene segment constellations (3,4). In 2014, along the Pacific flyway corridor in Canada and the United States, goose/Guangdong/1/96 (Gs/GD) lineage H5N8 virus was detected in wild waterfowl (5). The Gs/GD virus reassorted with NA lineage IAVs, subsequently resulting in devastating losses among commercial poultry flocks in Canada and the United States.
Wild bird–origin IAVs can breach the host species barrier and cause outbreaks involving several terrestrial mammals. After crossing the species barrier, some IAVs can become established in new hosts and circulate independently of their reservoir hosts. Different species of marine mammals are susceptible to IAV infection through exposure at haul-out sites, where they might comingle with wild birds, infected sympatric marine mammal species, terrestrial wildlife, including mink and river otters, and through direct exposure to infected humans or waterfowl in rehabilitation facilities (6). Previous outbreaks and individual case reports have shown that seals are susceptible to and have died as a result of infection with H10N7, H3N8, H7N7, H4N6, and other IAVs (6).
Figure 1
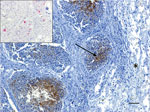
Figure 1. Immunohistochemistry testing for influenza A virus antigen in lung tissue of an adult male harbor seal, British Columbia, Canada. Viral antigen (arrow) was detected by immunohistochemistry within the bronchiolar-associated lymphoid...
A live adult male harbor seal weighing 49 kg washed ashore at Combers Beach near Tofino, British Columbia (BC), Canada, but died before he could be captured alive. On June 1, 2021, the seal was brought to the Animal Health Center Laboratory (Abbotsford, BC, Canada) for necropsy. The seal was in moderate body and fair postmortem condition. The most notable findings from gross examination were serosanguinous fluid within the thoracic cavity and focally extensive hemorrhage and edema from the skin to the visceral pleura along the right midlateral aspect of the thorax, with focal visceral to parietal pleural adhesion. Histopathology revealed necrotizing bronchitis and bronchiolitis, peribronchiolar lymphoid hyperplasia, alveolar histiocytosis, and perivascular lymphoplasmacytic cuffing. Immunohistochemistry testing identified IAV viral antigen within the bronchiolar-associated lymphoid tissue of 2 bronchioles in more severely affected areas of the lungs and in situ hybridization confirmed IAV RNA (Figure 1). Aerobic cultures of the lungs, hilar lymph node, brain, intercostal skeletal muscle, and small intestine yielded variable mixed growth of Streptococcus phocae and Serratia liquefaciens with no fungal growth from the lung. PCR testing of pooled tissues proved positive for consensus influenza virus and mollicutes (we confirmed Mycoplasma spp., but were unable to speciate) and negative for canine distemper virus.
We extracted RNA samples from the hilar lymph node, thymus, spleen, and lungs, and thoracic fluid tested positive on a real-time reverse transcription PCR assay based on IAV matrix (M) genes, as described elsewhere (7). We isolated all IAV PCR-positive samples in embryonated specific pathogen–free chicken eggs from lung tissue samples only. We amplified all 8 viral gene segments from original lung specimens and isolates as described elsewhere (8), then purified amplified reverse transcription PCR products using a QIAGEN QIAquick PCR purification kit (https://www.qiagen.com) and determined the concentration of the amplicons used for sequencing using an Invitrogen Qubit dsDNA BR assay kit (https://www.thermofisher.com) on the Qubit Fluorometer. We performed library prep using Illumina Nextera XT Library preparation kit (https://www.illumina.com) and sequencing using Illumina MiSeq, as described elsewhere (9). We assembled IAV full genome segments using DNASTAR SeqMan NGen software version 15.3.0 (https://www.dnastar.com).
Figure 2

Figure 2. Maximum-likelihood phylogenetic tree of influenza A virus subtype H10 hemagglutinin gene from an adult male harbor seal, British Columbia, Canada (red text), and reference sequences. Phylogenetic analyses were based on...
Full genome sequences of the virus obtained from the original lung tissue samples and isolates from lungs were 100% identical. Further analysis demonstrated that 4 gene segments, polymerase basic (PB) 1 and 2, polymerase acidic (PA), and M segments, originated from unknown NA lineage IAVs of wild bird origin. The remaining 4 gene segments, H, N, nucleoprotein (NP), and nonstructural (NS) were derived from EA lineage IAVs. We designated the virus A/harbor seal/British Colombia/OTH-52-1/2021(H10N7) and deposited sequences from the original sample and isolates into the National Center for Biotechnology Information genome database (https://www.ncbi.nlm.nih.gov/genome) under accession nos. OL336415 (PB2), OL336416 (PB1), OL336417 (PA), OL336418 (H), OL336419 (NP), OL336420 (N), OL336421 (M), and OL336422 (NS). We compared percentage similarity between all 8 gene segments and their associated proteins and closest matches in GenBank (Table). Phylogenetic analysis demonstrated that the H gene of the virus clades with EA lineage IAVs was circulating in wild birds (Figure 2).
Further analysis of the virus genome revealed that the H10N7 virus carries a glutamic acid (E) residue at the aa 627 site of PB2 determined to be associated with decreased replication in mammal cells using growth kinetics and with decreased virulence in mice using lethal dose challenge (10,11). We also observed an aspartic acid substitution at site aa 701, which has been associated with systemic replication and mortality in mice (11,12). In addition, this residue substitution in guinea pigs conferred efficient replication of the virus in the nose, trachea, and lungs (11). Key amino acid changes were also observed in NP and M segments. The presence of lysine at the aa 319 site in the NP gene segment has been implicated in species adaptation (12) and substitution in the A/seal/Mass/1/1980(H7N7) backbone conferred increased binding to importin alpha1. In the M protein, aspartate at aa 30 and alanine at aa 215 sites also increased virulence as indicated by the decreased postexposure survival rate in mice (13).
In our study of a harbor seal infected with a novel reassortment H10N7 IAV containing a unique constellation of NA and EA lineage gene segments, we found no conclusive evidence of where and when the reassortment occurred. However, the seal was recovered at a location within the Pacific avian flyway, where Gs/GD lineage H5N8 virus was detected in 2014 and later reassorted with NA lineage viruses to create novel reassortant H5N2 and H5N1 IAVs that were responsible for large outbreaks in domestic poultry in Canada and the United States (5). Harbor seals share habitat with seabirds and shorebirds, and the seal in our study may have been infected through spillover directly from infected birds as happened in cases reported elsewhere (6,8). In previous outbreaks in seals, the proximate cause of death was attributed to secondary or opportunistic Mycoplasma spp. (14), which were also detected in this case, or bacterial infection. Finally, because novel IAVs in marine mammals have been shown to be potentially zoonotic (15), IAV circulation among seals could have important public health implications for humans and other mammals.
Dr. Berhane is a virologist with the National Centre for Foreign Animal Disease, Canadian Food Inspection Agency in Winnipeg, Manitoba, Canada, and has appointments with the Department of Animal Science, University of Manitoba, Winnipeg, and Department of Veterinary Pathology, Western College of Veterinary Medicine, University of Saskatchewan, Saskatoon, Saskatchewan, Canada.
Acknowledgments
The authors thank Tamiko Hisanaga, Matt Suderman, Estella Moffat, and Katherine Handel for their technical assistance. The authors also thank staff from the necropsy, bacteriology, virology, and molecular diagnostics sections of the British Columbia Animal Health Centre for their technical assistance.
This study was supported by the Public Health Agency of Canada and Canadian Food Inspection Agency to support genetic and antigenic monitoring of influenza A virus in animals.
References
- Wu Y, Wu Y, Tefsen B, Shi Y, Gao GF. Bat-derived influenza-like viruses H17N10 and H18N11. Trends Microbiol. 2014;22:183–91. DOIPubMedGoogle Scholar
- Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56:152–79. DOIPubMedGoogle Scholar
- Huang Y, Wille M, Benkaroun J, Munro H, Bond AL, Fifield DA, et al. Perpetuation and reassortment of gull influenza A viruses in Atlantic North America. Virology. 2014;456-457:353–63. DOIPubMedGoogle Scholar
- Ramey AM, Reeves AB, Sonsthagen SA, TeSlaa JL, Nashold S, Donnelly T, et al. Dispersal of H9N2 influenza A viruses between East Asia and North America by wild birds. Virology. 2015;482:79–83. DOIPubMedGoogle Scholar
- Ramey AM, DeLiberto TJ, Berhane Y, Swayne DE, Stallknecht DE. Lessons learned from research and surveillance directed at highly pathogenic influenza A viruses in wild birds inhabiting North America. Virology. 2018;518:55–63. DOIPubMedGoogle Scholar
- Venkatesh D, Bianco C, Núñez A, Collins R, Thorpe D, Reid SM, et al. Detection of H3N8 influenza A virus with multiple mammalian-adaptive mutations in a rescued Grey seal (Halichoerus grypus) pup. Virus Evol. 2020;6:veaa 016.
- Weingartl HM, Berhane Y, Hisanaga T, Neufeld J, Kehler H, Emburry-Hyatt C, et al. Genetic and pathobiologic characterization of pandemic H1N1 2009 influenza viruses from a naturally infected swine herd. J Virol. 2010;84:2245–56. DOIPubMedGoogle Scholar
- Zhou B, Donnelly ME, Scholes DT, St George K, Hatta M, Kawaoka Y, et al. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol. 2009;83:10309–13. DOIPubMedGoogle Scholar
- Xu W, Weese JS, Ojkic D, Lung O, Handel K, Berhane Y. Phylogenetic inference of H3N2 canine influenza A outbreak in Ontario, Canada in 2018. Sci Rep. 2020;10:6309. DOIPubMedGoogle Scholar
- Bogs J, Kalthoff D, Veits J, Pavlova S, Schwemmle M, Mänz B, et al. Reversion of PB2-627E to -627K during replication of an H5N1 Clade 2.2 virus in mammalian hosts depends on the origin of the nucleoprotein. J Virol. 2011;85:10691–8. DOIPubMedGoogle Scholar
- Li J, Ishaq M, Prudence M, Xi X, Hu T, Liu Q, et al. Single mutation at the amino acid position 627 of PB2 that leads to increased virulence of an H5N1 avian influenza virus during adaptation in mice can be compensated by multiple mutations at other sites of PB2. Virus Res. 2009;144:123–9. DOIPubMedGoogle Scholar
- Gabriel G, Dauber B, Wolff T, Planz O, Klenk HD, Stech J. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc Natl Acad Sci U S A. 2005;102:18590–5. DOIPubMedGoogle Scholar
- Fan S, Deng G, Song J, Tian G, Suo Y, Jiang Y, et al. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology. 2009;384:28–32. DOIPubMedGoogle Scholar
- van Kuppeveld FJ, van der Logt JT, Angulo AF, van Zoest MJ, Quint WG, Niesters HG, et al. Genus- and species-specific identification of mycoplasmas by 16S rRNA amplification. Appl Environ Microbiol. 1992;58:2606–15. DOIPubMedGoogle Scholar
- van den Brand JMA, Wohlsein P, Herfst S, Bodewes R, Pfankuche VM, van de Bildt MWG, et al. Influenza A (H10N7) virus causes respiratory tract disease in harbor seals and ferrets. PLoS One. 2016;11:
e0159625 . DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleOriginal Publication Date: June 09, 2022
Table of Contents – Volume 28, Number 7—July 2022
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Stephen Raverty, Animal Health Center–Pathology, 1767 Angus Campbell Rd, Abbotsford, Vancouver, BC V3G 2M3, Canada
Top