Volume 11, Number 9—September 2005
Research
Molecular Epidemiology of SARS-associated Coronavirus, Beijing
Abstract
Single nucleotide variations (SNVs) at 5 loci (17564, 21721, 22222, 23823, and 27827) were used to define the molecular epidemiologic characteristics of severe acute respiratory syndrome–associated coronavirus (SARS-CoV) from Beijing patients. Five fragments targeted at the SNV loci were amplified directly from clinical samples by using reverse transcription–polymerase chain reaction (RT-PCR), before sequencing the amplified products. Analyses of 45 sequences obtained from 29 patients showed that the GGCTC motif dominated among samples collected from March to early April 2003; the TGTTT motif predominanted afterwards. The switch from GGCTC to TGTTT was observed among patients belonging to the same cluster, which ruled out the possibility of the coincidental superposition of 2 epidemics running in parallel in Beijing. The Beijing isolates underwent the same change pattern reported from Guangdong Province. The same series of mutations occurring in separate geographic locations and at different times suggests a dominant process of viral adaptation to the host.
Severe acute respiratory syndrome (SARS) is a new infectious disease that spread worldwide in early 2003, affecting >30 countries, with >8,098 cases and 774 deaths reported (1). Beijing, People's Republic of China, experienced the largest SARS outbreak in the world, with 2,523 cases and 181 deaths by June 12, 2003 (2,3). The epidemic occurred in 2 phases. The first phase began on March 5, 2003, and was caused by a patient who had been infected in Guangzhou and was involved in a superspreader event (SSE) in Beijing hospitals. Most patients in this period proved to be directly or indirectly linked with the index patient by traditional epidemiologic investigations. Molecular epidemiology, based on genome sequencing of the early isolates, also provided evidence that Beijing infections were closely related to those from the Guangdong epidemic (4). The second phase was marked by widespread transmission in healthcare facilities and communities, with incidence peaking in late April, followed by a dramatic decline in occurrence during the first week of May. The last probable case was noted on May 29, 2003 (5). During this phase, many case-patients had no apparent contact with SARS patients.
After the sequencing of the whole genome (6–9) information on viral strains from different geographic and temporal origins became available in GenBank. Comparative sequence analyses identified 5 loci, sequence variants of which segregated together as specific genotypic patterns, which could be used to define epidemic phases (10). All or some of the 5 loci were included in previous molecular epidemiologic studies (4,11–13), making them important genetic signatures to differentiate lineage-specific and temporal-specific patterns. In this study, we investigated the genetic variations of SARS-CoV in Beijing based on the 5-locus signature. Also, by sequence comparison among patients from 1 case cluster and different samples from 1 patient, the adaptable mutation of the virus in the host was further explored.
Participants
Study participants were recruited from 2 hospitals designated for SARS patients in Beijing. All of them fit the World Health Organization (WHO) case definition for probable SARS, i.e., temperature >38°C, cough or shortness of breath, new pulmonary infiltrates on chest radiograph, and a history of exposure to a SARS patient or of living in an area of on-going SARS transmission (14). After informed consent was obtained, epidemiologic and clinical data were collected from the participants by using a standard data collection form with interview and medical record review. The information obtained included the following items: age, sex, occupation, medical history, time and nature of exposure, symptoms and physical findings, laboratory tests at admission to hospital, and outcomes on discharge or transfer. Patients also provided clinical specimens (sputum and stool) for SARS-CoV detection by RT-PCR assay with specific primers (COR1, COR2) recommended by WHO. Only the patients with positive RT-PCR results were included in the study.
Laboratory Methods
Specimens were analyzed by using RT-PCR techniques. Briefly, total RNA was extracted by using the QIAamp virus RNA mini kit (Qiagen, Hilden, Germany) as instructed by the manufacturer. RNA was used to synthesize cDNA with the SuperScript II RNase H– reverse transcriptase system (Invitrogen, Carlsbad, CA, USA). Five sets of primers were used in nested PCR to amplify the fragments covering the 5-locus genetic signatures (17564, 21721, 22222, 23823, and 27827) (Table 1). Then, with the purified PCR products as templates and the second round primers as sequencing primers, the fragments were sequenced in ABI Prism 377 DNA sequencer (Applied Biosystems Inc, Foster City, CA, USA). Each PCR fragment was directly sequenced from both inward and outward directions, in duplicate.
All the original base data were processed for base calling, assembly, and editing by the SegMan II sequences analysis software of DNA Star package (DNASTAR, Madison, WI, USA). The comparisons with other sequences available from public database (GenBank) were made by using the default parameter of ClustalW (http://www.ebi.ac.uk/clustalw/). Single nucleotide variations (SNVs) were indicated, and the deduced amino acid changes were described.
A total of 160 samples (81 stools and 79 sputum samples) from 62 patients with positive results by RT-PCR were included this study. Of these, 45 samples (36 sputum samples and 9 stools) from 29 patients (17 men and 12 women, with a median age of 32 years) yielded amplicons for the 5 targeted loci (Table 2). The patients came from 2 SARS-designated hospitals in Beijing, with disease onset ranging from March to May, 2003. Four patients had serious conditions during hospitalization, including pulmonary aggravation requiring oxygen ventilation or transfer to an intensive care unit. No patient died.
The sequences of the 45 positive specimens were compared with SARS-CoV genome sequences available from the public database (GenBank). The sequence variants in 5 loci (17564, 21721, 22222, 23823, and 27827) defined 3 kinds of motifs: GGCTC, TGTTT, and GATTC (Table 2). In addition, 4 new SNVs were identified at nucleotides 17620, 22077, 22589, and 27749 in >1 patient. These variations appeared independently in several isolates, which indicates that they are not RT-PCR artifacts. None of them had been previously reported, with 3 nucleotide substitutions leading to amino acid changes (Table 3).
Twelve patients in this study belonged to a cluster. They derived from an SSE indirectly linked with the earliest SARS patients in Beijing. The first 2 patients of this cluster, who became ill on March 10 and 21, respectively, harbored the GGCTC motif. The remaining patients, who became ill from March 31 to May 4, showed the TGTTT motif. Among patients outside of the cluster, 5 of 6 patients with onset date before April had the GGCTC motif, while the TGTTT motif became predominant later (9 of 11 patients until May 12). A new motif, GATTC, was found in 2 patients outside the cluster. In addition, no intrapatient variation was observed in the 5 amplicons from specimens collected at different times or from different sources (sputum or stools).
The possible role of genetic mutations in patients' prognosis was also investigated. The presence of nucleotide substitution was compared between 2 groups of patients: 1 with good prognosis (absence of pulmonary aggravation; n = 25) and 1 with adverse outcome (pulmonary aggravation 8–12 days after onset of symptoms requiring oxygen ventilation or transfer to ICU; n = 4). No mutation was found associated with disease severity (Table 2).
Figure
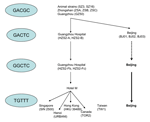
During the 2003 SARS epidemic, conventional epidemiologic investigation, aided by viral sequencing analysis, identified viral genetic signatures that are linked to geographic and temporal clusters of infection (4,10–12,15–18). Findings of these studies are summarized in the Figure, connecting the worldwide epidemic to a transmission event in hotel M in Hong Kong in late February 2003.
Beijing had experienced the SARS epidemic from March to June; however, only a few Beijing strains from the early epidemic have been analyzed in previous studies. Our study is the first to provide phylogenetic information on Beijing strains from the early and middle epidemic, as well as the late epidemic, by using the 5-locus motif of previous studies. The series of mutations in the 5-locus motif observed in Beijing followed the same path as isolates in Guangdong Province and the worldwide epidemic, i.e., the early introduction of GACTC motif was followed by transition to a GGCTC motif, before switching to a stable TGTTT motif. The observation of the same series of mutations occurring in 2 separate locations at different times suggests a dominant process of viral adaptation to the host. Moreover, this finding can expand our understanding of SARS-CoV response to selection pressures in humans, since early Beijing isolates (BJ01, BJ02, and BJ 03), which are traceable to Guangdong, underwent an independent selection process and would not be subject to the same sampling bias caused by superspreading events in Hong Kong isolates. The GGCTC→TGTTT switch was observed among patients belonging to the same cluster in this study, which rules out the possibility of the coincidental superposition of 2 epidemics (GGCTC and TGTTT) coexisting in Beijing.
The mutations involved in the GGCTC→TGTTT switch are responsible for amino acid changes in a nonstructural protein (17564, region Orf1b) in S protein (21721 and 22222) and in a noncoding region (27827, X3). We were not able to identify a correlation between these changes and the clinical status of patients. We did not find sequence variations in specimens obtained from the same patients either collected at different times or among different specimen types, which suggests that within-individual variations are rare in the partial genome of this study, although the phenomenon was described in a previous study (15). A new motif, GATTC, which represents a new transitional motif between GACTC and TGTTT, was described on 2 occasions in patients who were not part of the cluster. Similarly, 4 new SNVs were identified at nucleotides 17620, 22077, 22589, 27749.
In summary, this study confirms the evolution of SARS-CoV strains towards a TGTTT motif in positions 17564, 21721, 22222, 23823, and 27827 in Beijing, as was observed in Guangdong province before the hotel M outbreak in Hong Kong. Whether this motif is associated with higher transmission or virulence remains to be elucidated.
Dr Liu is an epidemiologist in the Department of Epidemiology, Beijing Institute of Microbiology and Epidemiology. Her primary research interests are molecular epidemiology and emerging infectious disease.
Acknowledgments
We thank Guo-Ping Zhao, Huai-Dong Song, and Guo-Wei Zhang for their assistance with this study.
This work was partly supported by the EC grant EPISARS (511063), the Programme de Recherche en Réseaux Franco-Chinois (P2R), the National Institutes of Health CIPRA Project (NIH U19 AI51915), and the National 863 Program of China (2003AA208406, 2003AA208412C).
References
- World Health Organization. SARS epidemiology to date [monograph on the Internet]. 2003. [cited 2003 Apr 11]. Available from: http://www.who.int/csr/sars/epi2003_04_11/en/
- World Health Organization. Multicentre Collaborative Network for Severe Acute Respiratory Syndrome (SARS) Diagnosis. A multicentre collaboration to investigate the cause of severe acute respiratory syndrome. Lancet. 2003;361:1730–3.PubMedGoogle Scholar
- World Health Organization. Cumulative number of reported probable cases of severe acute respiratory syndrome (SARS) [monograph on the Internet]. [cited 2003 Jul 11]. Available from: http://www.who.int/csr/sars/country/en/
- Guan Y, Peiris JS, Zheng B, Poon LL, Chan KH, Zeng FY, Molecular epidemiology of the novel coronavirus that causes severe acute respiratory syndrome. Lancet. 2004;363:99–104. DOIPubMedGoogle Scholar
- Pang X, Zhu Z, Xu F, Guo J, Gong X, Liu D, Evaluation of control measures implemented in the severe acute respiratory syndrome outbreak in Beijing, 2003. JAMA. 2003;290:3215–21. DOIPubMedGoogle Scholar
- Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–76. DOIPubMedGoogle Scholar
- Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–9. DOIPubMedGoogle Scholar
- Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–66. DOIPubMedGoogle Scholar
- Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–404. DOIPubMedGoogle Scholar
- Chinese SARS Molecular Epidemiology Consortium. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–9. DOIPubMedGoogle Scholar
- Zhong NS, Zheng BJ, Li YM, Poon LLM, Xie ZH, Chan KH, Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People's Republic of China, in February, 2003. Lancet. 2003;362:1353–8. DOIPubMedGoogle Scholar
- Ruan YJ, Wei CL, Ee AL, Vega VB, Thoreau H, Su ST, Comparative full-length genome sequence analysis of 14 SARS coronavirus isolates and common mutations associated with putative origins of infection. Lancet. 2003;361:1779–85. DOIPubMedGoogle Scholar
- Tsui SK, Chim SS, Lo YM; Chinese University of Hong Kong Molecular SARS Research Group. Coronavirus genomic-sequence variations and the epidemiology of the severe acute respiratory syndrome. N Engl J Med. 2003;349:187–8. DOIPubMedGoogle Scholar
- World Health Organization. Case definitions for surveillance of severe acute respiratory syndrome (SARS). [cited 2003 Apr 29]. Available at: http://www.who.int/csr/sars/casedefinition/en
- Xu DP, Zhang Z, Chu Fl, Li Y, Jin L, Zhang L, Genetic variation of SARS coronavirus in Beijing hospital. Emerg Infect Dis. 2004;10:789–94.PubMedGoogle Scholar
- Yeh SH, Wang HY, Tsai CY, Kao CL, Yang JY, Liu HW, Characterization of severe acute respiratory syndrome coronavirus genomes in Taiwan: molecular epidemiology and genome evolution. Proc Natl Acad Sci U S A. 2004;101:2542–7. DOIPubMedGoogle Scholar
- Tsang KW, Ho PL, Ooi GC, Yee WK, Wang T, Chan-Yeung M, A cluster of cases of severe acute respiratory syndrome in Hong Kong. N Engl J Med. 2003;348:1977–85. DOIPubMedGoogle Scholar
- Wang Z, Li L, Luo Y, Zhang J, Wang M, Cheng S, Molecular biological analysis of genotyping and phylogeny of severe acute respiratory syndrome associated coronavirus. Chin Med J (Engl). 2004;117:42–8.PubMedGoogle Scholar
Figure
Tables
Cite This ArticleTable of Contents – Volume 11, Number 9—September 2005
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Wu-Chun Cao, Beijing Institute of Microbiology and Epidemiology, State Key Laboratory of Pathogen and Biosecurity, P.R. China; fax: 86-10-63812060
Top