Volume 14, Number 5—May 2008
Letter
Hantavirus Outbreak, Germany, 2007
To the Editor: Hantavirus disease (for review see [1]) has been reportable in Germany since 2001, according to the Federal Infection Protection Act. In this country, Puumala virus (PUUV) causes most clinical hantavirus cases, although Dobrava-Belgrade virus and Tula virus also circulate (1). From 2001 through 2006, an average of ≈220 cases were reported per year (incidence 0.267/100,000) with a maximum of 448 cases in 2005. In contrast, 1,687 cases were reported in 2007 (2). Whereas in 2005 the highest incidence of infection was in metropolitan areas (3), the current outbreak is focused in the rural areas in southern and western Germany. Clinical case-patients exhibit key characteristics of hantavirus disease (nephropathia epidemica): acute high fever; pain in the back, head, and/or abdomen; proteinuria; rise of serum creatinine; thrombocytopenia; and renal failure (1). The outbreak provided considerable numbers of clinical samples from the viremic phase and thus has enabled a molecular epidemiologic analysis of the circulating virus.
At the National Consultation Laboratory for Hantavirus Infections (Berlin), we received early-phase serum specimens from the outbreak regions for confirmation assays. In enzyme immunoassays and Western blot tests (4), 80 samples from patients during the early clinical phase were positive for PUUV-specific immunoglobulin (Ig) M antibodies. All IgM data were accompanied by simultaneous or subsequent detection of PUUV-specific IgG. The samples were screened for hantavirus RNA by reverse transcription–PCR (RT-PCR) (5). Of the 80 early-phase serum samples, 42 (53%) were RT-PCR positive. For a subset of 14 of the 42 samples, a 557-nt segment of the nucleocapsid (S) gene underwent nucleotide sequence analysis as described previously (6).
Figure
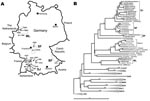
Figure. A) Map of Germany showing origins of viral sequences from the 2007 outbreak. H, sequences of human origin; M, sequences of rodent origin (Myodes glareolus). Dotted circles mark the outbreak regions...
The Figure, panel A, shows a map of Germany with the residences of those patients from whom virus sequences were amplified (marked by letter H in front of the specimen number). In the phylogenetic analysis, despite a substantial evolutionary distance to PUUV strains from other parts of Europe, the virus sequences unambiguously grouped within the PUUV species (Figure, panel B). The few previously known human PUUV sequences from individual clinical case-patients in Germany, “Berkel” from Munsterland (7) and “Heidelberg” from a region located between Swabian Jura and Spessart Forest (8), as well as human-derived strains from a small 2004 outbreak in the Bavarian Forest (6), were included in this analysis. The results showed a clustering of the new viral sequences strictly according to residential areas of the patients, forming the following 4 clades: Swabian Jura (SJ), Spessart Forest (SF), Munsterland (ML), and Bavarian Forest (BF). Two different single sequences, Karlsruhe (from a region in northwestern Swabian Jura) and Essen (in southern Munsterland), represent 2 putative additional lineages.
Most sequences in this study were obtained from Swabian Jura, the region with the highest illness rate of the outbreak (incidence 32.9/100,000). The Swabian Jura was previously identified as a hantavirus-endemic area characterized by higher seroprevalence rates in the population compared with the rest of Germany (9). Sequence alignments within this clade showed a nucleotide sequence diversity of up to 5.5%. Within the BF clade, the diversity is up to 4%. However, between the 4 phylogenetic clades mentioned above (SJ, SF, ML, and BF), a sequence variability of 12%–18% was found.
The natural reservoir of PUUV is the bank vole, Myodes glareolus; the virus is transmitted to humans by the aerosolized excreta of these rodents (1). Sequence comparisons showed a tight correlation between human- and rodent-derived PUUV sequences obtained from the same regional provenance (nucleotide identity >98%) and high variability of sequences originating from different geographic regions (nucleotide identity ≈85%). Neighbor-joining analyses confirmed the direct clustering of human- and rodent-derived sequences in the different phylogenetic clades (Figure, panel B).
In this study we focused on the analysis of a 557-nt S-segment region. For more detailed studies, analysis of the complete S and M sequences of the virus strains will be necessary. Nevertheless, our results demonstrate a high variability among the German PUUV strains but a strong clustering of viral sequences of human and rodent origin in the same geographic region. The diversity of the PUUV clusters suggests their separate evolutionary history in the different regions of Germany. In contrast, within these particular geographic areas, only slight sequence differences were found in longitudinal analysis over several years. This conclusion is supported by the novel human Waldkirchen sequence (H72), which is almost identical to the BF strains from 2004 (6,10) and the similarity of newly derived human sequences from Munsterland (H208, H303) to the Berkel strain from 1994 (7). The molecular characterization of the viral sequences of patient and rodent origin from the outbreak areas demonstrates that PUUV is the causative agent of the current outbreak.
Acknowledgments
We thank Jens Jacob and many other colleagues for help in the collection and primary preparation of human or rodent specimens.
This study was supported by Deutsche Forschungsgemeinschaft (grant KR1293/2-4) and Paul und Ursula Klein-Stiftung.
References
- Kruger DH, Ulrich R, Lundkvist A. Hantavirus infections and their prevention. Microbes Infect. 2001;3:1129–44. DOIPubMedGoogle Scholar
- Robert Koch Institute. Berlin, Germany. Epidemiologisches Bulletin. 2008;(13) [cited 2008 <ar 28]. Available from http://www.rki.de
- Abu Sin M, Stark K, van Treek U, Dieckmann H, Uphoff H, Hautmann W, Risk factors for hantavirus infection in Germany, 2005. Emerg Infect Dis. 2007;13:1364–6.PubMedGoogle Scholar
- Meisel H, Wolbert A, Razanskiene A, Marg A, Kazaks A, Sasnauskas K, Development of novel immunoglobulin G (IgG), IgA, and IgM enzyme immunoassays based on recombinant Puumala and Dobrava hantavirus nucleocapsid proteins. [PMID 17021245]. Clin Vaccine Immunol. 2006;13:1349–57. DOIPubMedGoogle Scholar
- Kramski M, Meisel H, Klempa B, Kruger DH, Pauli G, Nitsche A. Detection and typing of human pathogenic hantaviruses by real-time reverse transcription-PCR and pyrosequencing. Clin Chem. 2007;53:1899–905. DOIPubMedGoogle Scholar
- Schilling S, Emmerich P, Klempa B, Auste B, Schnaith E, Schmitz H, Hantavirus disease outbreak in Germany: limitations of routine serological diagnostics and clustering of virus sequences of human and rodent origin. J Clin Microbiol. 2007;45:3008–14. DOIPubMedGoogle Scholar
- Pilaski J, Feldmann H, Morzunov S, Rollin PE, Ruo SL, Lauer B, Genetic identification of a new Puumala virus strain causing severe hemorrhagic fever with renal syndrome in Germany. J Infect Dis. 1994;170:1456–62.PubMedGoogle Scholar
- Bahr U, Zeier M, Muranyi W. Characterization of a new Puumala virus genotype associated with hemorrhagic fever with renal syndrome. Virus Genes. 2006;33:229–34. DOIPubMedGoogle Scholar
- Zoller L, Faulde M, Meisel H, Ruh B, Kimmig P, Schelling U, Seroprevalence of hantavirus antibodies in Germany as determined by a novel recombinant enzyme immunoassay. Eur J Clin Microbiol Infect Dis. 1995;14:305–13. DOIPubMedGoogle Scholar
- Essbauer S, Schmidt J, Conraths FJ, Friedrich R, Koch J, Hautmann W, A new Puumala hantavirus subtype in rodents associated with an outbreak of Nephropathia epidemica in South-East Germany in 2004. Epidemiol Infect. 2006;134:1333–44. DOIPubMedGoogle Scholar
Figure
Cite This Article1These authors contributed equally to this article.
2Current affiliation: Bernhard-Nocht-Institute for Tropical Medicine, Hamburg, Germany.
Related Links
Table of Contents – Volume 14, Number 5—May 2008
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Detlev H. Kruger, Institute of Virology–School of Medicine (Charité), Helmut Ruska House, Humboldt University, Berlin D-10098, Germany;
Top