Volume 17, Number 5—May 2011
Research
Molecular Epidemiology of Oropouche Virus, Brazil
Abstract
Oropouche virus (OROV) is the causative agent of Oropouche fever, an urban febrile arboviral disease widespread in South America, with >30 epidemics reported in Brazil and other Latin American countries during 1960–2009. To describe the molecular epidemiology of OROV, we analyzed the entire N gene sequences (small RNA) of 66 strains and 35 partial Gn (medium RNA) and large RNA gene sequences. Distinct patterns of OROV strain clustered according to N, Gn, and large gene sequences, which suggests that each RNA segment had a different evolutionary history and that the classification in genotypes must consider the genetic information for all genetic segments. Finally, time-scale analysis based on the N gene showed that OROV emerged in Brazil ≈223 years ago and that genotype I (based on N gene data) was responsible for the emergence of all other genotypes and for virus dispersal.
Oropouche virus (OROV) is one of the most common orthobunyaviruses (family Bunyaviridae, genus Orthobunyavirus) (1) and is the causative agent of Oropouche fever in humans, which is clinically characterized as an acute febrile disease (2). The first isolation of OROV was reported in Trinidad and Tobago in 1955, when the virus was isolated from the blood of a febrile patient and from a pool of Coquillettidia venezuelensis mosquitoes (3). OROV was described in Brazil in 1960, when it was isolated from a sloth (Bradypus tridactylus) captured near a forested area during construction of the Belem–Brasilia highway and from a pool of Ochlerotatus (Ochlerotatus) serratus mosquitoes, captured near the same site (4).
Since the first isolation of OROV, >30 outbreaks have been reported in Brazil, Peru, Panama, and Trinidad and Tobago during 1960–2009. At least half a million persons are estimated to have been infected (5,6).
Similar to the genomes of other orthobunyaviruses, the OROV genome comprises 3 single-stranded negative-sense RNA segments—large, medium, and small. The large RNA segment encodes a large protein that has RNA polymerase activity for transcription and replication of genomic RNA segments. The medium segment encodes a precursor polyprotein, which gives rise to the viral surface glycoproteins (Gc and Gn) and to a nonstructural protein NSM. The small RNA encodes a structural nucleocapsid (N) protein, as well as a smaller nonstructural protein (NSS) in overlapping reading frames (1). Studies of the molecular biology of the OROV small RNA segment have suggested its monophyletic origin and the existence of at least 3 genotypes (I, II, and III) (7). Recently, genotype III was isolated from a wild vertebrate host (Callithrix sp.) in southeastern Brazil, suggesting possible dispersion of the virus to susceptible and populated areas in Brazil (8). Further molecular analyses that used OROV strains recovered during outbreaks in Pará State during 2003–2007 demonstrated the association of at least 2 different genotypes (I and II) with Oropouche fever cases in the area (9,10).
In this study, we describe new information regarding the molecular epidemiology of OROV. This information will help clarify the evolution, dispersal, and genotyping classification of this human pathogen in the Brazilian Amazon region.
Virus Strains
The OROV strains used in this study (Table A1) were relatively low-passage isolates obtained from the virus collection of the Department of Arbovirology and Hemorrhagic Fevers, Evandro Chagas Institute (Ananindeua, Brazil). These strains corresponded to viruses recovered from different hosts and geographic locations that were isolated during 1960–2009.
Virus Culture and RNA Extraction
Viruses were propagated in monolayer cultures of Vero cells. After 75% of cells showed cytopathic effects, the supernatants of infected cell cultures were collected. RNA extraction was conducted by using a commercial kit (QIAmp Viral RNA Mini Kit; QIAGEN, Valencia, CA, USA) according to the manufacturer’s instructions.
Reverse Transcription–PCR and Nucleotide Sequencing
For the synthesis and amplification of the OROV small RNA, medium RNA, and large RNA cDNA (cDNA), a 1-step reverse transcription–PCR (RT-PCR) was conducted by using a combination of specific-segment sets of the following primers: small RNA (NORO5: AAAGAGGATCCAATAATGTCAGAGTTCATTT; ORO N3: GTGAATTCCACTATATGCCAATTCCGAATT), medium RNA (Gn15S: GGCAACAAACAGTGACAAT and Gn659R: CTATGTTAACGCACATTGCT), and large RNA (LOROF: CCGAAACAAACAAAAACAAT; and large RNA (LOROF: CCGAAACAAACAAAAACAAT and LOROR: GGATGAGTAAGCAATTCTGG) (7). Amplicon lengths were expected to be 693 bp, 644 bp, and 634 bp for small RNA, medium RNA, and large RNA, respectively. The RT-PCR products were visualized onto 1.2% agarose gel stained with ethidium bromide (0.5 μg/mL). Amplicons were sequenced by using the same primers applied for the RT-PCR cycling and the ABI PRISM Dye Terminator Kit (Applied Biosystems, Foster City, CA, USA) by using the dideoxyribonucleotide chain terminator method (11). The ABI 3130 capillary automated sequencer (Applied Biosystems) was used to obtain the sequence. Both cDNA strands were sequenced from at least 3 RT-PCR products.
Sequence Analysis and Phylogeny
Sequences obtained for the N (complete), Gn, and large (partial) genes were first inspected in quality by the SeqMan LaserGene package (DNA STAR, Madison, WI, USA) and then used for multiple sequencing alignments with other OROV sequences available in GenBank (www.ncbi.nlm.nih.gov/genbank). The genetic divergence for each gene was determined by using MEGA4 software (12) based on the dataset generated by the alignments. Confidence interval for inclusion into a given phylogenetic group was estimated according to the mean of genetic divergence calculated for the known OROV genotypes (I, II, and III) and used as a criterion for searching other genotype groups.
The phylogenetic analysis was performed by comparing the 66 entire N genes and 36 partial Gn genes and large sequences of Brazilian OROV strains, respectively, with homologous sequences obtained from other OROVs isolated from different regions of Central and South America, periods of time, and source of isolation (Table 1). Phylogenetic trees were constructed by using the neighbor-joining (13), maximum-likelihood, and maximum-parsimony methods in the PAUP 4.0 software (14) as described (8). Bayesian and time-scaled (chronologic) analyses also were conducted as described by Rodrigues et al. (15). Sequences obtained from the OROV isolates were deposited in GenBank (Table, GenBank accession numbers of previously sequenced OROV and other Simbu group virus strains; Table A1).
Evaluation of RNA Segment Topologies
To evaluate the topologies presented by the different RNA segments, we used 36 OROV strains for which all 3 segments were sequenced. The evaluation was performed by using the Kishino-Hasegawa method (16), comparing a topology generated for a given RNA segment with the other segments. We considered p values <0.01 significant.
Genetic Variability of OROV
The nucleotide sequences obtained for the studied strains were 693 nt (231 aa), 644 nt (214 aa), and 634 nt (211 aa) in length for N, Gn, and large genes, respectively. The multiple sequencing analysis of the new 66 full-length OROV N (small RNA) and for the 36 partial Gn (medium RNA) and large RNA gene sequences showed high nucleotide and amino acid identities (>90%). The mean of genetic divergence among the N gene nucleotide sequence was ≈6.8%. Genetic distances (nucleotide sequence) within the 3 well-established genotypes (I, II, and III) ranged from 3% between genotypes I and II to 4.4% between genotypes I and III (mean 3.5%) and were used as a confidence value for inclusion within a given genotype. On the basis of this criterion, a fourth group was established, and a genetic divergence ranging from 5.3% with genotype I to 6.8% with genotype III (mean 5.8%) was determined. The mean of genetic divergence among the 4 OROV lineages was 4.6% (Table 1).
Regarding the Gn gene nucleotide sequences, the analysis showed values of genetic divergence of 0.9%–9.5% (mean 6.5%). In contrast to the N gene sequences, for the Gn gene partial sequences, 3 lineages were identified, showing an intergroup divergence of 4.5% (between groups I and II) to 7.2% (between groups I and III) (mean 5.7%), which was used as a confidence value for group inclusion or exclusion (Table 1).
For the polymerase gene nucleotide sequences (large RNA), genetic divergence was 0.1%–0.8% (mean 0.5%). Only 2 large RNA segments were distinguished into groups (Table 1).
Phylogeny and Time-scaled Analysis
Figure 1

Figure 1. Phylogenetic analysis between Oropouche virus (OROV) (N gene: 693 nt) and homologue sequences of different viruses that belong to the Simbu group. AINOV, Aino virus; AKAV, Akabane virus; TINV, Tinaroo virus;...
Regardless of the method used, the trees were similar in topology, showing high support values (bootstrap, likelihood, or posterior probability values). The Bayesian method showed high support values (>0.90) and was therefore used to represent the final tree. As previously reported (7–10), the comparative phylogeny that used the entire N gene sequences (96 strains; Table A1) confirmed the monophyletic origin of OROV in comparison with other Simbu group viruses (Figure 1),
Figure 2
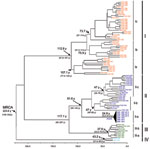
Figure 2. Phylogenetic tree based on the complete nucleotide (nt) sequence of the N gene (693 nt) of 96 Oropouche virus (OROV) strains isolated from different hosts, locations, and periods. The main phylogenetic...
The 4 major phylogenetic groups depicted (I–IV) corresponded to 4 distinct genotypes (Figure 2 [Bayesian method]). Genotype I included the Brazilian strains isolated in the states of Acre, Amazonas, Maranhão, Tocantins, and Pará, as well as strains from Trinidad and Tobago. Three subgenotypes were described: Ia, Ib, and Ic (Figure 1). Genotype II grouped strains obtained during outbreaks in the states of Amapá, Pará, and Rondônia in Brazil and the strains from Peru. Three subgenotypes also were assigned to this group (II a, II b, and II c). Genotype III was formed by strains isolated in the Brazilian states of Acre, Minas Gerais, and Rondônia, and the isolates from Panama showing 2 distinct sublineages: the subgenotypes II a and III b. Finally, genotype IV included the Brazilian strains isolated in Amazonas State, Brazil (Figure 2).
Chronologic analysis was used to investigate the emergence period of OROV in the Americas. The nucleotide substitution rate that determined the 96 OROV N gene sequences was 3.7 × 10–4 substitutions per site per year and was used to estimate the divergence dates among the strains. The emergence of the most recent common ancestor (MRCA) for OROV in the Americas was estimated to have occurred ≈223 years ago (95% highest probability density [HPD] 148–342 years) from the location where the other parental viruses for the different genotypes (I, II, III, and IV) emerged (Figure 2). The estimated emergence dates suggest that genotype I was the first genotype that emerged ≈112 years ago (95% HPD 95–189 years). Genotype II emerged ≈91 years ago (95% HPD 59–144 years) and originated from strains isolated in the states of Pará and Rondônia, and strains recently isolated in Amapá State, in 2009. Genotype III was estimated to have originated 37 years ago (95% HPD 33–70 years) and probably evolved in Rondônia State 33 years ago (95% HPD 29–58 years), and other Amazonian states, such as Acre and Pará, emerging almost simultaneously in Panama 32 years ago (95% HPD 22–45 years) and, more recently, in Minas Gerais State. Genotype IV emerged in Amazonas State ≈43 years ago (95% HPD 31–56 years; Figure 2).
Evaluation of RNA Segment Topologies
Figure 3

Figure 3. Phylogenetic analysis of 36 Oropouche virus strains: A) N gene (693 nt), B) Gn gene (644 nt), and C) large (L) gene (634 nt), showing different topologies. Bootstrap values obtained by...
Trees generated from entire N and partial Gn and large gene sequences obtained for 36 OROV strains demonstrated different topologies. By using all phylogenetic methods, we found differences in virus clustering. For the small RNA, 4 distinct groups were identified: group I (20 strains), group II (9 strains), group III (5 strains), and group IV (2 strains). For the medium RNA, 3 groups were assigned and distributed as follows: group I (28 strains), group II (4 strains), and group III (4 strains). The large RNA depicted only 2 major groups, including 32 strains in group I and 4 strains in group II (Figure 3). Maximum likelihood was used to analyze these competing small, medium, and large segment topologies by using the Kishino-Hasegawa test. Sequence evolution models were optimized by applying all genome segments and using the competing topologies. Regardless of which model was selected, each topology generated by using maximum parsimony and neighbor-joining methods with a given genome segment was significantly more likely than the competing topology generated by using the other genome segment (likelihood probability between S and M topologies = 0.00005623; likelihood probability between S and L topologies = 0.000354664; likelihood probability between M and L topologies = 0.00043154; p<0.001).
Geographic Dispersion of OROV Genotypes
Figure 4
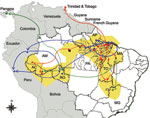
Figure 4. Geographic dispersion of Oropouche virus (OROV) genotypes in South America during 1955–2009 based on data from the N gene. Yellow shading, coverage area of OROV in Brazil; red line, dispersion route...
On the basis of results obtained for the N gene data by time-scaled analysis (evolutionary rate and emergence date) and epidemiologic data association (date and place of isolation), the possible dispersal event could be predicted for the distinct OROV genotypes in the Americas (Figure 4). Genotype I (dispersion route in red), originally isolated in Brazil in the municipality of Ipixuna, Pará State (BR 010 Highway, km 94), possibly dispersed continuously toward distinct directions: initially to several municipalities in western Pará, and simultaneously in Trinidad and Tobago. Later, genotype I moved toward the states of Amazonas and Acre and, more recently, to the eastern Amazon region including Pará, Maranhão, and Tocantins States. Genotype II (dispersion route in dark blue), apparently emerged simultaneously in the states of Amapá, Pará, and Rondônia, as well as in Peru, and dispersed in these places, emerging in the municipality of Mazagão, Amapá State, in 2009. Genotype III (dispersion route in green), emerged in Rondônia State, moving toward Panama and the states of Acre and Maranhão. From Maranhão, a new route led genotype III to the Minas Gerais State. Genotype IV (black dot in Manaus), apparently more ancient than genotype III, emerged in the city of Manaus, Amazonas State, and it has not apparently dispersed from there (Figure 4).
The molecular epidemiology of OROV has been extensively studied on the basis of genetic data generated for the small RNA segment, and the data have provided information about the genetic diversity of OROV and geographic distribution in countries in which the virus is endemic, such as Brazil, Peru, and Trinidad and Tobago (7–10,17). The analysis of additional 66 gene sequences of the entire N and partial Gn and Gc provided a better understanding of the molecular epidemiology of OROV in Brazil. In our analysis, distinct phylogenetic groups were observed when the different RNA segments were analyzed. In case of the small RNA, 4 major groups were found, including the 3 genotypes previously described (7–10,17). Although a fourth genetic lineage has been well established by the small RNA phylogeny (strains AM 01 and AM 03), the topologies depicted by the medium RNA and large RNA sequences did not support this result. Maximum likelihood analyses were used to test these competing small, medium, and large segment topologies by using the Kishino-Hasegawa test. Evolution models were optimized for all 3 genome segment sequences and by using the competing topologies. Regardless of which model was selected, each topology generated by using maximum parsimony and neighbor-joining methods with a given RNA segment was significantly more likely than the competing topology generated by the other genome segment (p<0.001) (Figure 3). These results ensured that the testing topologies obtained for each RNA segment differed significantly, which suggests that each OROV RNA segment had a different evolutionary history and probably contributes to the genetic variability of the virus.
The assessment of additional genetic data for the small RNA segment contributed substantially to the understanding of the emergence of the virus, geographic distribution, and dispersal events. On the basis of chronologic dating of the N gene, epidemiologic data, and lineage definition (genotypes I–IV), we were able to elucidate the possible origin of OROV in the Americas (Figure 2, Figure 4). In contrast to information about the event in Trinidad and Tobago in 1955 that was associated with the first description of the Oropouche fever case, molecular data provided by the small RNA sequences indicated that OROV emerged in South America, more precisely in Pará State (strains PA 01–PA 05) in northern Brazil, ≈89 years ago, and then in Trinidad and Tobago probably through humans carrying the virus during the viremic phase or through illegal shipment of wild animals, as has been suggested for yellow fever virus (18).
The dispersal history of OROV strains is initially associated with genotype I, more precisely with the subgenotype Ia, isolated from wild animals and humans during epidemics in Pará State, during the 1960s–1970s. Their dispersion routes were simultaneously west to east in the Amazon toward Acre State (subgenotype Ib) from 1988 to 1994 and, more recently, in a vast area in Pará State and in Manaus, Amazonas State, at the end of the 1990s and the beginning of the 2000s.
Regarding genotype II, the most probable origins were in eastern Pará (Porto de Moz) toward Iquitos, Peru (subgenotype IIb), and from Iquitos toward Ariquemes, Rondônia State (subgenotype IIc), where the virus probably then dispersed to Madre de Dios in Peru and to Pará State. The origin of subgenotype IIa, which is represented by the strains recently associated with the epidemic in Mazagão, Amapá State, in the beginning of 2009 (P.F.C. Vasconcelos et al., pers. comm.) is probably related to a common ancestor that evolved independently from other subgenotypes (IIb and IIc) over time and probably emerged in the Amazon ≈24 years ago.
The existence of genetic data for a single genotype III Brazilian strain isolated in Minas Gerais State, southeastern Brazil (8), limited our ability to study its origin and evolutionary aspects. With the identification of other genotype III strains in Brazil, isolated in the states of Rondônia (Ariquemes and Machadinho d’Oeste), Acre (Xapuri), and Maranhão (Porto Franco), we were able to make inferences about the most possible dispersion route. In fact, it constitutes a complex dynamics of evolutionary origin between subgenotypes IIIa (predominantly from Brazil) and IIIb (predominantly from Panama). In this context, genotype III probably originated from the sublineage IIIa, which was isolated in Ariquemes, Rondônia State, from which the sublineage IIIb ancestor has segregated independently, leading to the emergence of strains in Chame and San Miguelito, Panama.
In a more detailed view, the subgenotype IIIa found in Ariquemes, Rondônia State, had its initial dispersion to a neighboring municipality (Machadinho d’Oeste), subsequently to Porto Franco in Maranhão State, and finally to Arinos, Minas Gerais State. Although Minas Gerais State is geographically distant from the official OROV-endemic area, the virus may have been introduced through Maranhão State by the intense traffic of humans from Maranhão to other states and regions in Brazil.
In Minas Gerais, OROV has been maintained in silent cycles, probably because of inadequate epidemiologic conditions, such as the high density of Culicoides paraensis mosquitoes in urban areas, a limiting factor for an epidemic cycle deflagration. Furthermore, the sporadic detection of OROV was recently reported in Acre State (19); these reports confirmed that the virus actually circulates silently in the Brazilian Amazon, as suggested by Azevedo et al. (9), and can be transported by viremic patients and human carriers of subclinical illness from region to region within the country. This approach should result in stronger data when new isolates are sequenced in other OROV-endemic countries because limited information about dispersal of OROV in Peru, Panama, and Trinidad and Tobago does not infer a more robust analysis.
In conclusion, even with the limited data obtained in this study from other OROV-endemic countries, we were able to reach a more complete understanding of the molecular epidemiology of the virus, and we provided evidence of which distinct genes (N, Gn/Gc, and L) are under different selective evolutionary pressures in nature. We also observed the great genetic diversity of OROV, the description of a new genotype IV, the complex dynamics of evolution, and viral dispersal. Finally, our findings suggest the necessity of obtaining genetic data regarding full-length sequencing of different OROV strains (medium and large segments) to elucidate the correct genotype classification and to improve the molecular diagnostics of this human pathogen in Latin America.
Dr Baldez Vasconcelos is a researcher at the Instituto Evandro Chagas, Ananindeua, Pará, Brazil, specializing in molecular biology of arboviruses. Her research interests include the molecular epidemiology of dengue virus, yellow fever virus, OROV, and other human arboviruses.
Acknowledgments
We thank Basílio Silva Buna, Creuza Lima Carvalho, Jefferson Amaral Buna, Jonas Morais, Luiz Roberto Oliveira Costa, and Osvaldo Vaz da Silva for their technical work and Nelson Veiga Gonçalves for assistance with Figure 4.
This research was partially supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (former Conselho Nacional de Pesquisa) (grants 300460/2005-8, 483453/2007-2, and 302987/2008-8), IEC/SVS.
References
- Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. Bunyaviridae. In: Fauquet CM, Mayo MA, Maniloff J, Desselberguer U, Ball LA, editors. Virus taxonomy classification and nomeclature of viruses. Eighth report of the International Committee on Taxonomy of Viruses. Virology Division. International Union of Microbiological Societies. San Diego (CA): Elsevier; 2005. p. 695–9.
- Le Duc JW, Pinheiro FP. Oropouche fever. In: Monath TP, editor. The arboviruses: epidemiology and ecology. Boca Raton (FL): CRC Press; 1988. p. 1–14.
- Anderson CR, Spence L, Downs WG, Aitken THG. Oropouche virus: a new human disease agent from Trinidad. West Indies. Am J Trop Med Hyg. 1961;10:574–8.PubMedGoogle Scholar
- Pinheiro FP, Pinheiro M, Bensabath G, Causey OR, Shope RE. Epidemia de vírus Oropouche em Belém. Rev Serv Esp Saúde Públ. 1962;12:15–23.
- Pinheiro FP, Travassos da Rosa APA, Travassos da Rosa JF, Ishak R, Frietas RB, Gomes ML, Oropouche virus. I. A review of clinical, epidemiological and ecological findings. Am J Trop Med Hyg. 1981;30:149–60.PubMedGoogle Scholar
- Pinheiro FP, Travassos da Rosa APA, Vasconcelos PFC. Oropouche fever. In: Feigin RD, editor. Textbook of pediatric infectious diseases. Philadelphia: Saunders; 2004. p. 2418–23.
- Saeed MF, Wang H, Nunes MRT, Vasconcelos PFC, Weaver SC, Shope RE, Nucleotide sequences and phylogeny of the nucleocapsid gene of Oropouche virus. J Gen Virol. 2000;81:743–8.PubMedGoogle Scholar
- Nunes MRT, Martins LC, Rodrigues SG, Chiang JO, Azevedo RS, Travassos da Rosa AP, Oropouche virus isolation, southeast Brazil. Emerg Infect Dis. 2005;11:1610–3.PubMedGoogle Scholar
- Azevedo RSS, Nunes MRT, Chiang JO, Bensabath G, Vasconcelos HB, Pinto AYN, Reemergence of Oropouche fever, northern Brazil. Emerg Infect Dis. 2007;13:912–5.PubMedGoogle Scholar
- Vasconcelos HB, Azevedo RSS, Casseb SM, Nunes-Neto JP, Chiang JO, Cantuária PC, Oropouche fever epidemic in northern Brazil: epidemiology and molecular characterization of isolates. J Clin Virol. 2009;44:129–33. DOIPubMedGoogle Scholar
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–7. DOIPubMedGoogle Scholar
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. DOIPubMedGoogle Scholar
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstruction phylogenetic trees. Mol Biol Evol. 1987;4:406–25.PubMedGoogle Scholar
- Swofford DL. PAUP: Phylogenetic analysis using parsimony and other methods, version 4. Sunderland (MA): Sinauer Associates; 2002.
- Rodrigues SG, Nunes MRT, Casseb SMM, Prazeres ASC, Rodrigues DSG, Silva M, Molecular epidemiology of the Saint Louis encephalitis virus in the Brazilian Amazon: genetic divergence and dispersal. J Gen Virol. 2010;91:2420–7. DOIPubMedGoogle Scholar
- Goldman N, Anderson JP, Rodrigo AG. Likelihood-based test of topologies in phylogenetics. Syst Biol. 2000;49:652–70. DOIPubMedGoogle Scholar
- Nunes MRT, Vasconcelos HB, Medeiros DBA, Rodrigues SG, Azevedo RS, Chiang JO, A febre do Oropouche: uma revisão dos aspectos epidemiológicos e moleculares na Amazônia brasileira. Cad Saúde Colet. 2007;13:303–18.
- Vasconcelos PFC, Bryant JE, Travassos da Rosa APA, Tesh RB, Rodrigues SG, Barrett ADT. Genetic divergence and dispersal of yellow fever virus, Brazil. Emerg Infect Dis. 2004;10:1578–84.PubMedGoogle Scholar
- Bernardes-Terzian AC, Bronzoni RVM, Drumond BP, Silva-Nunes M, Silva NS, Ferreira MU, Sporadic Oropouche virus infection, Acre, Brazil. Emerg Infect Dis. 2009;15:348–50. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 17, Number 5—May 2011
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Pedro F.C. Vasconcelos, Instituto Evandro Chagas, Rodovia BR-316, KM 7, CEP 66030-000, Ananindeua, Pará, Brazil
Top