Volume 5, Number 1—February 1999
Synopsis
Using Monoclonal Antibodies to Prevent Mucosal Transmission of Epidemic Infectious Diseases
Abstract
Passive immunization with antibodies has been shown to prevent a wide variety of diseases. Recent advances in monoclonal antibody technology are enabling the development of new methods for passive immunization of mucosal surfaces. Human monoclonal antibodies, produced rapidly, inexpensively, and in large quantities, may help prevent respiratory, diarrheal, and sexually transmitted diseases on a public health scale.
In 1975, Köhler and Milstein noted that monoclonal antibodies (MAbs) "...could be valuable for medical and industrial use" (1). Since then, the use of MAbs has become routine in the research and diagnostic laboratory, but antibodies have yet to be used to their maximum potential in medical and public health applications. Two recent reviews of the therapeutic use of antibodies suggest that systemically administered antibodies may play an important role in treating infections by drug-resistant pathogens as well as pathogens for which no antimicrobial drugs are available (2,3). However, the greatest potential for MAbs probably lies in prevention since antibodies are in general more effective for prophylaxis than for therapy (3,4). From a public health perspective, prevention is especially important (5). In particular, direct application of MAbs to mucosal surfaces blocks the entry of pathogens into the body.
We review here the evidence of antibody efficacy in preventing disease and recent advances that have facilitated the development of MAbs for mucosal applications in humans. Finally, we consider the public health potential of topical delivery of MAbs for preventing mucosal transmission of infections.
Vaccines that stimulate systemic immunity can prevent systemic disease, but generally fail to prevent mucosal disease. Vaccines that stimulate active mucosal immunity have demonstrated good efficacy in animal models, but with few exceptions (polio and influenza vaccines), have not been as effective as they could be in humans. Some of the discrepancies between study results in animals and humans are probably due to a failure of studies in animals to model immune evasion strategies of pathogens (6) that occur in humans. These strategies include rapid evolution of variable strains (7), pathogens that coat themselves with host antigens (8), and pathogens that are transmitted to a new host by hiding inside cells shed by the infected host (cell vectors) (9). Furthermore, most vaccines successful in stimulating mucosal immunity in animals contain irritating adjuvants or attenuated pathogens, which are generally considered unacceptable for use in humans; vaccines with human-safe adjuvants have not generated high concentrations of protective antibody in the mucosa. Current research is investigating improved immunogens, delivery vehicles, and adjuvants, as well as exploring the best inductive sites for generating a protective mucosal immune response at a specific mucosal surface (10).
Figure 1
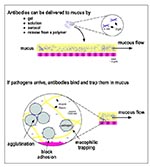
Figure 1. Topical delivery of pathogen-specific MAbs can protect the mucosal epithelium. (Top) Protective MAbs (in this figure, secretory immunoglobulin A; SIgA) can be topically applied to the mucosa in various ways. (Bottom)...
In contrast to vaccines, passive immunizations can deliver protective levels of antibodies immediately and directly to the susceptible mucosal surface (Figure 1-top). Also, with passive mucosal immunization, it may be possible to defeat some key immune evasion strategies by using antibodies directed against host cell vectors, host antigens that coat the pathogen, or receptors used by pathogens to enter target cells (11). In addition, new methods for the sustained release of antibodies offer the possibility of long-term protection (12).
The first use of immune serum for preventing disease by passive immunization was reported more than 100 years ago by von Behring and Kitasato (13). Subsequently, systemic passive immunization with antibodies has been proven effective in preventing many diseases. By binding to a pathogen, systemically delivered antibodies can inhibit attachment to and fusion with target cells, inhibit internalization by target cells, inhibit uncoating inside a cell, aggregate pathogens thereby preventing them from reaching target cells, interact with complement to lyse the pathogen, induce phagocytosis of the pathogen, and cause killer cells to lyse the pathogen by antibody-dependent cellular cytotoxicity (14). Table 1 lists the highest efficacy reported for systemically delivered antibodies in preventing disease in mammalian species and against a wide range of pathogens that infect humans. No antiviral treatments are available for most viruses listed in the table, yet antibodies can prevent the diseases caused by all of these viruses.
Although less studied than systemic passive immunization, the prophylactic use of mucosal antibodies predates the therapeutic use of immune sera. Antibodies delivered in mother's milk have been protecting the gastrointestinal tract of nursing infants since the mammary gland first evolved approximately 50 million years ago. Most infections begin in mucosal surfaces (approximately 400 m2 in an adult human); supplementing the antibody repertoire in a mucous secretion (Figure 1-top) thus offers an effective method for protecting a mucosal surface against pathogens to which the host has not been exposed or become immune. In addition to the protective mechanisms described above, antibodies delivered to mucosal surfaces can trap pathogens in the mucous gel, make them mucophilic, and prevent their diffusion and motility (Figure 1-bottom); as a result, pathogens trapped in mucus are shed from the body with the normal flow of mucous secretions or are digested if these secretions enter the digestive tract (61-63). Topical passive immunization of mucosa can block transmission of bacteria, viruses, fungi, and parasites that infect humans (Table 2).
The predominant (and perhaps the most appropriate for mucosal delivery) antibody isotype on most human mucosal surfaces is secretory immunoglobulin A (SIgA); efficient methods for producing SIgA have been reported (82,83). SIgA, a tetravalent dimer of monomeric IgA associated with two polypeptides (joining chain and secretory component), is especially stable and well suited to function in the enzymatically hostile environment that prevails at mucosal surfaces (84). SIgA, the least phlogistic class of antibody (84), is the least likely to induce inflammatory responses that can make it easier for toxins and pathogens to breach the mucosal surface. Immune exclusion of antigens, enzymes, and toxins has been repeatedly demonstrated in vivo, and protection generally correlates with levels of SIgA antibodies in the relevant mucous secretions. Finally, the protective role of SIgA has been demonstrated in many systems (85).
Generating High-Affinity Human MAbs
Figure 2
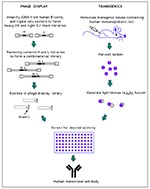
Figure 2. Generation of human monoclonal antibodies. (Phage display) Heavy and light chain cDNA isolated from human B-cells is used to generate a combinatorial library in which random heavy (H) and light chain...
Since the advent of cloning of human antibodies from combinatorial libraries constructed from seropositive persons (86,87), generation of fully human MAbs against human pathogens has become routine (Figure 2) (88). For example, from a single bone marrow donor, human MAbs were prepared against HIV, respiratory syncytial virus
(RSV), cytomegalovirus, herpes simplex virus types 1 and 2, varicella zoster virus, and rubella virus (88). MAbs can even be obtained from naive libraries prepared from unexposed persons (if the library has a large enough repertoire) (89); therefore, antibodies against pathogens lethal to humans can be generated. Alternatively, human MAbs can be generated by traditional immunization of commercially available mice that have been genetically engineered to contain human immunoglobulin loci in their germline (Figure 2) (90,91).
Dramatic enhancement of the affinity of an mAb has been demonstrated by molecular biologic techniques in which mutants of an antibody are generated and then screened for higher affinity or higher neutralization activity (93-95). For example, the affinity of one anti-HIV mAb has been enhanced 420-fold, and this matured antibody neutralizes more HIV strains than the original mAb (94). Furthermore, expressing a mAb as a multivalent isotype, such as SIgA or IgM, can dramatically enhance the potency of an antibody by increasing the avidity (96) or agglutination activity (14). For example, an anti-Escherichia coli IgM was 1,000-fold more effective in protecting neonatal rats than its class-switched IgG (both in vitro and in vivo) (41). From a commercial standpoint, a 1,000-fold increase in avidity could translate into a 1,000-fold decrease in dose and subsequent cost. Also, a large dose of a highly potent mAb can substantially increase the duration of protection (97).
Production Systems
MAbs have traditionally been produced in cell culture and have been prohibitively expensive for most preventive uses. Over the years, however, the cost has continually dropped; MAbs are now being produced in cell culture for $200 to $1,000 per gram (98,99). Production of MAbs has recently been reported in both transgenic plants and animals (82,100,101). Both of these systems are expected to lower costs dramatically. Indeed, transgenic plants can be scaled up in agricultural fields to produce tons of "plantibody," and plant-produced antibody is predicted to cost less than U.S. $1/g (102). The actual cost, however, will remain unknown until large-scale batches are produced, purified, and formulated in accordance with Good Manufacturing Practices.
Safety and Regulatory Status
More than 80 MAbs are now in clinical trials (most for cancer imaging or therapy) and more than one quarter of these are in phase III trials (103). Few safety problems have been reported for systemic applications; antibodies are now considered "biotechnology-derived pharmaceuticals" by the U.S. Food and Drug Administration (FDA)—enabling a more straightforward regulatory process than in the past (92,104). Even though MAbs have often been evaluated for systemic applications, only recently have they been evaluated in humans for mucosal applications. This new interest in mucosal antibodies may be partially due to the increasing recognition of the importance of mucosal immunity. Only two clinical trials have evaluated topically delivered MAbs: intranasally delivered anti-RSV in infants at high risk (105) and orally delivered anti-Streptococcus mutans in adults (106); no major adverse effects were reported in these studies.
Safety concerns, such as peptide and glycosylation immunogenicity, are important when MAbs are delivered systemically but are likely to be of less concern when MAbs are applied to the mucosa, a surface that has evolved to interact with the external environment. Indeed, antibodies delivered to the lumen of a mucosal surface have minimal interaction with circulating immune cells. Although proteins, and even antibodies, can be absorbed through mucosal surfaces (107,108), generally only small quantities are absorbed (109,110). The inability of SIgA to activate complement by the classic pathway is likely involved in maintaining the integrity of mucosal surfaces (63); therefore, SIgA may be preferable to IgG or IgM for many mucosal applications.
The FDA "Points to Consider" for characterization of antibodies produced in cell-culture and transgenic animals (111) are better defined than for characterization of antibodies produced in transgenic plants; however, plant-derived antibodies are free of animal viruses and may therefore not require rigorous viral inactivation processing steps. In addition, although glycosylation patterns of MAbs produced in mammalian cell-culture and transgenic animals are closer phylogenetically to humans than glycosylation patterns in plants, given our repeated exposure to plant sugars in food and personal care products, it is unlikely that any of these patterns are novel to human immune systems (112). In fact, in a recently completed clinical trial with repeated applications of plant-produced antibody for the prevention of oral colonization by S. mutans, no safety problems were encountered, nor were there any detectable human anti-plant antibody responses (113).
Selection for resistant organisms by widespread and repeated use of antibiotics is a serious health concern (60). Drug-resistant strains of a wide variety of pathogens have already been reported (Table 1). Antibiotic or antiviral treatment of infected persons in which pathogens are actively replicating provides a strong evolutionary selection process for developing drug-resistant pathogens. In contrast, MAbs are less likely to create resistant organisms when used in a preventive context at a mucosal surface against a pathogen that is not yet actively replicating. Even if a systemic infection does occur during topical use of MAbs, resistant organisms will likely not be created since the pathogen will not be replicating and evolving in the presence of the mAb applied to the mucosal surface. This is in marked contrast to the settings in which antibiotics and antiviral drugs select for resistant strains (60). If MAbs are used frequently on a population level, the risk of selecting for resistant organisms may increase. When the emergence of resistant strains is of particular concern, the tendency to select mAb-resistant organisms could be minimized by using cocktails of mucosal antibodies directed at multiple antigenic targets (2,114). Because new MAbs can be produced with a rapid turnaround time (discussed below), the emergence of an antibody-resistant strain could be countered by producing a new mAb directed toward the mutated epitope or another antigenic target of the resistant strain. Indeed, the flexibility of the antibody structure to create a virtually inexhaustible repertoire of antigen binding specificities suggests that immunoglobulins evolved in part as a means to cope rapidly with new pathogens.
Turnaround Time for Developing a New mAb
Since human MAbs can be identified quickly by cloning variable regions from specific antigen-binding human lymphocytes (115) or panning combinatorial libraries (87), antibodies could be used as a rapidly developed method for defending against new pathogens. The time required for collecting lymphocytes from a seropositive person, screening for an appropriate antibody, cloning, and expressing the antibody in culture in a well-equipped laboratory is 1 to 3 months; quantities sufficient for protecting persons at high risk or those at the focal point of an outbreak could be available in fewer than 6 months. High-capacity production in quantities sufficient for broad public health application could be available in several years, assuming that the safety of antibodies as a class of molecules is established and an infrastructure is in place for producing these antibodies. While in rare instances vaccines can be developed this quickly (e.g., the 1976 influenza vaccine [5]), new vaccines, antibiotics, and antiviral therapies usually take considerably longer to develop. Moreover, even though passive immunization may require repeated applications, MAbs delivered to a mucosal surface can provide immediate protection against infection.
From a public health perspective, MAbs are most promising for preventing gastrointestinal, respiratory, and reproductive tract infections. These infections cause almost 11 million deaths annually worldwide, accounting for more than 50% of the deaths caused by communicable diseases and 22% of deaths by all causes (116). Sexually transmitted diseases (STDs) accounted for 87% of all cases reported among the top ten most frequently reported diseases in 1995 in the United States; more than 12 million Americans are infected with STDs each year at an estimated annual cost of more than $12 billion (117).
If a track record of safety and efficacy can be achieved, mucosal antibodies will probably be most useful as over-the-counter products that could reach populations not well integrated into the health-care system. The condom, a nonmedical over-the-counter personal protection product, has played an important preventive role in the HIV epidemic. Personal protection provided by over-the-counter antibody-based technology could play a similar role in future emerging disease epidemics.
Diarrheal Disease
Studies in animal models have demonstrated that orally delivered antibodies were 100% effective in preventing rotavirus (70) and cholera (79) infections. In humans, orally delivered bovine antibodies were 100% effective in preventing rotavirus (118), enterogenic E. coli (74), Shigella infection (76), and necrotizing enterocolitis (119).
For orally delivered MAbs, digestive degradation is a potential concern. However, significant levels of functional antibody survive treatment with pepsin at pH 2 or with a pool of pancreatic enzymes at pH 7.5 in vitro (120). In addition, most ingested IgA in milk survives passage through the gastrointestinal tract of infants (121); intact antibody delivered orally with an antacid survived passage through the gastrointestinal tract of adults (74,76). Assuming that a 10-mg dose of antibody is protective (i.e., assuming that the mAb is only 100-fold more potent than polyclonal preparations [118]), the production costs for the amount of plantibody needed for 100 days of protection could be approximately one cent (102).
Since diarrheal diseases are most prevalent in developing countries, preventive strategies must be extremely inexpensive; therefore, MAbs produced in plants or in the milk of animals are likely most suitable for these countries. Because of the speed with which MAbs pass through the gastrointestinal tract, antibodies delivered orally will need to be delivered frequently, perhaps more than once a day. In endemic-disease regions, MAbs could be delivered orally as a supplement with food or water.
Respiratory Disease
Animal studies have demonstrated the efficacy of nasal delivery of antibodies for the prevention of RSV infection (71) and influenza (68). In one study, topical application was approximately 100 times more effective than systemic delivery (122). Another study found an anti-RSV mAb (MEDI-493) to be approximately 100 times more effective than an equal quantity of a polyclonal preparation (32). These results suggest that 10,000 times less anti-RSV mAb would be required for topical applications than for systemically delivered polyclonal preparations. Protective systemic doses of MEDI-493 are approximately 100 mg (15 mg/kg) (32), so <1 mg might suffice for protection if this mAb were applied topically. Intranasally applied mAb has a residence half-time of a little under one day in the monkey (71), suggesting that once-a-day applications that deliver several-fold more than a protective dose can provide continuous protection. MAbs for protecting the respiratory tract could be delivered in nose drops or by aerosol once a day to those at particular risk (e.g., infants and the elderly during influenza season) or to everyone living near the epicenter of an epidemic.
STDs
With the exception of hepatitis B, no vaccines are available for the prevention of STDs (Table 3). Until effective and safe vaccines are developed, vaginal delivery of a cocktail of anti-STD pathogen MAbs might make an effective new method for broad spectrum protection against STDs (11). In animal models, MAbs have been shown to protect against transmission of C. albicans, C. trachomatis, HSV, HIV, and syphilis (Tables 1, 2) (11). Antibodies have been delivered experimentally to the vagina in solution, gels, and more recently, by sustained release devices for long-term delivery of protective MAbs (123,124). Antibodies were found to be stable when stored in seminal fluid or cervical mucus for 48 hours at 37°C (125); no significant inactivation occurred over the pH range of the human vagina (pH 4 to 7) for at least 24 hours at 37°C (Zeitlin et al., unpub. obs.). Since the effective half-life of antibodies applied topically depends on the turnover time of mucus, a single vaginal application may thus provide protection for at least 1 day, and probably several days (97). If so, passive immunization of the vagina may extend protection to the occasional days when the user forgets to apply the mAb. Considering there are an estimated 5 billion acts of sexual intercourse per year in the United States (11), large-scale production of MAbs in plants may offer the best system for the low costs needed for such a public health initiative. In addition, because the most common class of infection in the first month of life is primarily caused by STD pathogens present in the birth canal (126), the same mucosal antibodies could be used in a predelivery cervicovaginal lavage or applied to newborns' eyes for studies in the prevention of ophthalmia neonatorum. Indeed, in some cultures the mother's colostrum, a fluid rich in SIgA, is applied to the newborns' eyes (127).
In animal models and human studies, antibodies have been shown to prevent a wide variety of infectious human diseases. Recent advances allow development of a new era of mucosal mAb-based products. These advances include the development of combinatorial libraries for rapid selection of human MAbs, the ability to increase dramatically the potency of a specific mAb, and the marked reduction in the cost of cell-culture—produced MAbs as well as the ability to produce MAbs inexpensively and at high capacity in transgenic animals and plants. In addition, since MAbs can be developed considerably more rapidly than most vaccines and antimicrobial drugs, MAbs may prove useful for combating emerging pathogens. Mucosal infections account for a large percentage of infectious disease-related illness and deaths; hence topical passive immunization with MAbs may offer a new opportunity for improving public health. Finally, many of the remaining safety issues regarding the human use of mucosal MAbs are likely to be addressed by clinical trials now under way.
Dr. Zeitlin is a research scientist at ReProtect, LLC. His interests focus on the development of monoclonal antibodies for contraception and the prevention of sexually transmitted diseases.
Acknowledgment
The authors thank the Rockefeller Foundation Bellagio Study and Conference Center and Drs. Polly F. Harrison, Mich B. Hein, and Thomas R. Moench for their review of drafts.
References
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–7. DOIPubMedGoogle Scholar
- Casadevall A, Scharff M. Return to the past: the case for antibody-based therapies in infectious diseases. Clin Infect Dis. 1995;21:150–61.PubMedGoogle Scholar
- Casadevall A. Antibody-based therapies for emerging infectious diseases. Emerg Infect Dis. 1996;2:200–8. DOIPubMedGoogle Scholar
- Cross A. Intravenous immunoglobulins to prevent and treat infectious diseases. In: Atassi MZ, GS Bixler GSJ, editors. Immunobiology of proteins and peptides VIII. New York: Plenum Press; 1995.
- Lederberg J, Shope R, Oaks S. Emerging Infections. Washington: National Academy Press; 1992.
- Mims C, Dimmock N, Nash A, Stephen J. Mims' pathogenesis of infectious disease. 4th ed. San Diego: Academic Press; 1995.
- Mo H, Stamatatos L, Ip JE, Barbas CF, Parren PWHI, Burton DR, Human immunodeficiency virus type 1 mutants that escape neutralization by human monoclonal antibody IgG1b12. J Virol. 1997;71:6869–74.PubMedGoogle Scholar
- Mandrell RE, Apicella MA. Lipo-oligosaccharides (LOS) of mucosal pathogens: molecular mimicry and host-modification of LOS. Immunobiology. 1993;187:382–402.PubMedGoogle Scholar
- Anderson D, Yunis E. "Trojan horse" leukocytes in AIDS. N Engl J Med. 1983;309:984–5.
- Ogra P. Mucosal immunoprophylaxis: an introductory overview. In: Kiyono H, Ogra P, McGhee J, editors. Mucosal vaccines. New York: Academic Press; 1996. p. 1-14.
- Cone RA, Whaley KJ. Monoclonal antibodies for reproductive health: Part I. Preventing sexual transmission of disease and pregnancy with topically applied antibodies. Am J Reprod Immunol. 1994;32:114–31.PubMedGoogle Scholar
- Saltzman WM. Antibodies for treating and preventing disease: the potential role of polymeric controlled release. Crit Rev Ther Drug Carrier Syst. 1993;10:111–42.PubMedGoogle Scholar
- Silverstein A. History of immunology. In: Paul W, editor. Fundamental immunology. 2nd ed. New York: Raven Press Ltd.; 1989.
- Dimmock N. Neutralization of animal viruses. Curr Top Microbiol Immunol. 1993;183:1–149.PubMedGoogle Scholar
- Igarashi A, Fukuoka T, Fukai K. Passive immunization of mice with rabbit antisera against Chikungunya virus and its components. Biken J. 1971;14:353–5.PubMedGoogle Scholar
- CytoGam package insert. Gaithersburg (MD): MedImmune Inc.
- Men RH, Bray M, Lai CJ. Carboxy-terminally truncated dengue virus envelope glycoproteins expressed on the cell surface and secreted extracellularly exhibit increased immunogenicity in mice. J Virol. 1991;65:1400–7.PubMedGoogle Scholar
- Mikhailov VV, Borisevich IV, Chernikova NK, Potryvaeva NV, Krasnianskii VP. The evaluation in hamadryas baboons of the possibility for the specific prevention of Ebola fever. Vopr Virusol. 1994;39:82–4.PubMedGoogle Scholar
- Arikawa J, Yao JS, Yoshimatsu K, Takashima I, Hashimoto N. Protective role of antigenic sites on the envelope protein of Hantaan virus defined by monoclonal antibodies. Arch Virol. 1992;126:271–81. DOIPubMedGoogle Scholar
- Eis-Hübinger A, Schmidt D, Schneweis K. Anti-glycoprotein B monoclonal antibody protects mice against genital herpes simplex virus infection by inhibition of virus replication at the inoculated mucous membranes. J Gen Virol. 1993;74:379–85. DOIPubMedGoogle Scholar
- Atherton SS. Protection from retinal necrosis by passive transfer of monoclonal antibody specific for herpes simplex virus glycoprotein D. Curr Eye Res. 1992;11:45–52. DOIPubMedGoogle Scholar
- Safrit JT, Fung MS, Andrews CA, Braun DG, Sun WN, Chang TW, hu-PBL-SCID mice can be protected from HIV-1 infection by passive transfer of monoclonal antibody to the principal neutralizing determinant of envelope gp120. AIDS. 1993;7:15–21. DOIPubMedGoogle Scholar
- Stapleton JT. Passive immunization against hepatitis A. Vaccine. 1992;10:S45–7. DOIPubMedGoogle Scholar
- McGory RW, Ishitani MB, Oliveira WM, Stevenson WC, McCullough CS, Dickson RC, Improved outcome of orthotopic liver transplantation for chronic hepatitis B cirrhosis with aggressive passive immunization. Transplantation. 1996;61:1358–64. DOIPubMedGoogle Scholar
- Okuno Y, Matsumoto K, Isegawa Y, Ueda S. Protection against the mouse-adapted A/FM/1/47 strain of influenza A virus in mice by a monoclonal antibody with cross-neutralizing activity among H1 and H2 strains. J Virol. 1994;68:517–20.PubMedGoogle Scholar
- Jahrling PB, Peters CJ. Passive antibody therapy of Lassa fever in cynomolgus monkeys: importance of neutralizing antibody and Lassa virus strain. Infect Immun. 1984;44:528–33.PubMedGoogle Scholar
- Giraudon P, Wild T. Correlation between epitopes on hemagglutinin of measles virus and biological activities: passive protection by monoclonal antibodies is related to their hemagglutination inhibiting activity. Virology. 1985;144:46–58. DOIPubMedGoogle Scholar
- Chanock R, Crowe J, Murphy B, Burron D. Human monoclonal antibody Fab fragments cloned from combinatorial libraries: potential usefulness in prevention and/or treatment of major human viral diseases. Infect Agents Dis. 1993;2:118–31.PubMedGoogle Scholar
- Dietzschold B, Kao M, Zheng YM, Chen ZY, Maul G, Fu ZF, Delineation of putative mechanisms involved in antibody-mediated clearance of rabies virus from the central nervous system [published erratum appears in Proc Natl Acad Sci U S A 1992;89:9365]. Proc Natl Acad Sci U S A 1992;89:7252-6. 630.
- Sherry B, Li XY, Tyler KL, Cullen JM, Virgin HW IV. Lymphocytes protect against and are not required for reovirus-induced myocarditis. J Virol. 1993;67:6119–24.PubMedGoogle Scholar
- Niklasson BS, Meadors GF, Peters CJ. Active and passive immunization against Rift Valley fever virus infection in Syrian hamsters. APMIS. 1984;92:197–200.
- MedImmune reports fourth set of clinical results evaluating MEDI-493 [press release]. Gaithersburg (MD): MedImmune; May 6, 1997.
- The PREVENT Study Group. Reduction of respiratory syncytial virus hospitalization among premature infants and infants with bronchopulmonary dysplasia using respiratory syncytial virus immune globulin prophylaxis. Pediatrics. 1997;99:93–9. DOIPubMedGoogle Scholar
- Neumann-Haefelin D, Neumann-Haefelin C, Petersen EE, Luthardt T, Hass R. Passive immunization against rubella: studies on the effectiveness of rubella-immuno-globulin after intranasal infection with rubella vaccination virus. Dtsch Med Wochenschr. 1975;100:177–81. DOIPubMedGoogle Scholar
- Brunell P, Ross A, Miller L. B K. Prevention of varicella by zoster immune globulin. N Engl J Med. 1969;280:1191–4.PubMedGoogle Scholar
- Danes L, Hruskova J. Efficiency testing of passive immunization against Venezuelan equine encephalomyelitis in mice. Acta Virol. 1969;13:554–6.PubMedGoogle Scholar
- Johnson RC, Kodner C, Russell M. Passive immunization of hamsters against experimental infection with the Lyme disease spirochete. Infect Immun. 1986;53:713–4.PubMedGoogle Scholar
- Sato Y, Sato H. Further characterization of Japanese acellular pertussis vaccine prepared in 1988 by 6 Japanese manufacturers. Tokai J Exp Clin Med. 1988;13:79–88.PubMedGoogle Scholar
- Kaukoranta-Tolvanen SE, Laurila AL, Saikku P, Leinonen M, Laitinen K. Experimental Chlamydia pneumoniae infection in mice: effect of reinfection and passive immunization. Microb Pathog. 1995;18:279–88. DOIPubMedGoogle Scholar
- Cotter TW, Meng Q, Shen ZL, Zhang YX, Su H, Caldwell HD. Protective efficacy of major outer membrane protein-specific immunoglobulin A (IgA) and IgG monoclonal antibodies in a murine model of Chlamydia trachomatis genital tract infection. Infect Immun. 1995;63:4704–14.PubMedGoogle Scholar
- Raff HV, Bradley C, Brady W, Donaldson K, Lipsich L, Maloney G, Comparison of functional activities between IgG1 and IgM class-switched human monoclonal antibodies reactive with group B streptococci or Escherichia coli K1. J Infect Dis. 1991;163:346–54.PubMedGoogle Scholar
- Drabick J, Narayanan R, Williams J, LeDuc J, Nacy C. Passive protection of mice against lethal Francisella tularensis (live tularemia vaccine strain) infection by the sera of human recipients of the live tularemia vaccine. Am J Med Sci. 1994;308:83–7. DOIPubMedGoogle Scholar
- Shigeoka AO, Pincus SH, Rote NS, Hill HR. Protective efficacy of hybridoma type-specific antibody against experimental infection with group-B Streptococcus. J Infect Dis. 1984;149:363–72.PubMedGoogle Scholar
- Schreiber JR, Barrus V, Cates KL, Siber GR. Functional characterization of human IgG, IgM, and IgA antibody directed to the capsule of Haemophilus influenzae type B. J Infect Dis. 1986;153:8–16.PubMedGoogle Scholar
- Hayatsu E, Kawakubo Y, Yayoshi M, Araake M, Wakai M, Yoshida A, Immunological responses of hamsters in the acquired immune state to Mycoplasma pneumoniae infection. Microbiol Immunol. 1981;25:1255–63.PubMedGoogle Scholar
- Brodeur BR, Larose Y, Tsang P, Hamel J, Ashton F, Ryan A. Protection against infection with Neisseria meningitidis group B serotype 2b by passive immunization with serotype-specific monoclonal antibody. Infect Immun. 1985;50:510–6.PubMedGoogle Scholar
- Harmon RC, Rutherford RL, Wu HM, Collins MS. Monoclonal antibody-mediated protection and neutralization of motility in experimental Proteus mirabilis infection. Infect Immun. 1989;57:1936–41.PubMedGoogle Scholar
- Fisher MW. A polyvalent human gamma-globulin immune to Pseudomonas aeruginosa: passive protection of mice against lethal infection. J Infect Dis. 1977;136:S181–5.PubMedGoogle Scholar
- Svenson SB, Nurminen M, Lindberg AA. Artificial Salmonella vaccines: O-antigenic oligosaccharide-protein conjugates induce protection against infection with Salmonella typhimurium. Infect Immun. 1979;25:863–72.PubMedGoogle Scholar
- Adamus G, Mulczyk M, Witkowska D, Romanowska E. Protection against keratoconjunctivitis shigellosa induced by immunization with outer membrane proteins of Shigella spp. Infect Immun. 1980;30:321–4.PubMedGoogle Scholar
- Scott DF, Best GK, Kling JM, Thompson MR, Adinolfi LE, Bonventre PF. Passive protection of rabbits infected with toxic shock syndrome-associated strains of Staphylococcus aureus by monoclonal antibody to toxic shock syndrome toxin 1. Reviews of Infectious Diseases 1989;11:S214-7; discussion S217-8.
- Swendsen CL, Johnson W. Humoral immunity to Streptococcus pneumoniae induced by a pneumococcal ribosomal protein fraction. Infect Immun. 1976;14:345–54.PubMedGoogle Scholar
- Azadegan AA, Schell RF, LeFrock JL. Immune serum confers protection against syphilitic infection on hamsters. Infect Immun. 1983;42:42–7.PubMedGoogle Scholar
- Motin V, Nakajima R, Smirnov G, Brubaker R. Passive immunity to Yersiniae mediated by anti-recombinant V antigen and protein A-V antigen fusion peptide. Infect Immun. 1994;62:4192–201.PubMedGoogle Scholar
- Friedlander AM, Welkos SL, Worsham PL, Andrews GP, Heath DG, Anderson GW Jr, Relationship between virulence and immunity as revealed in recent studies of the F1 capsule of Yersinia pestis. Clin Infect Dis. 1995;21:S178–81.PubMedGoogle Scholar
- Tavares D, Ferreira P, Vilanova M, Videira A, Arala-Chaves M. Immunoprotection against systemic candidiasis in mice. Int Immunol. 1995;7:785–96. DOIPubMedGoogle Scholar
- Nussbaum G, Yuan R, Casadevall A, Scharff MD. Immunoglobulin G3 blocking antibodies to the fungal pathogen Cryptococcus neoformans. J Exp Med. 1996;183:1905–9. DOIPubMedGoogle Scholar
- Diggs CL, Hines F, Wellde BT. Plasmodium falciparum: passive immunization of Aotus lemurinus griseimembra with immune serum. Exp Parasitol. 1995;80:291–6. DOIPubMedGoogle Scholar
- Johnson AM, McDonald PJ, Neoh SH. Monoclonal antibodies to Toxoplasma cell membrane surface antigens protect mice from toxoplasmosis. J Protozool. 1983;30:351–6.PubMedGoogle Scholar
- Garrett L. The coming plague. New York: Penguin Books; 1995.
- Kraehenbuhl JP, Neutra MR. Molecular and cellular basis of immune protection of mucosal surfaces. Physiol Rev. 1992;72:853–79.PubMedGoogle Scholar
- Cone R. Mucus. In: Ogra PL, Mestecky J, Lamm ME, Strober W, McGhee JR, Bienenstock J, editors. Mucosal immunology. 2nd ed. New York: Academic Press; 1999.
- Lamm M. Interaction of antigens and antibodies at mucosal surfaces. Annu Rev Microbiol. 1997;51:311–40. DOIPubMedGoogle Scholar
- Whaley KJ, Zeitlin L, Barratt RA, Hoen TE, Cone RA. Passive immunization of the vagina protects mice against vaginal transmission of genital herpes infections. J Infect Dis. 1994;169:647–9.PubMedGoogle Scholar
- Zeitlin L, Whaley KJ, Sanna PP, Moench TR, Bastidas R, De Logu A, Topically applied human recombinant monoclonal IgG1 antibody and its Fab and F(ab')2 fragments protect mice from vaginal transmission of HSV-2. Virology. 1996;225:213–5. DOIPubMedGoogle Scholar
- Zeitlin L. Topical methods for preventing genital herpes infection in the mouse. Reproductive biology. [dissertation]. Baltimore: The Johns Hopkins University; 1996.
- Jakeman K, Smith H, Sweet C. Mechanism of immunity to influenza: maternal and passive neonatal protection following immunization of adult ferrets with a live vaccinia-influenza virus hemagglutinin recombinant but not with recombinants containing other influenza virus proteins. J Gen Virol. 1989;70:1523–31. DOIPubMedGoogle Scholar
- Tamura S, Funato H, Hirabayashi Y, Suzuki Y, Nagamine T, Aizawa C, Cross-protection against influenza A virus infection by passively transferred respiratory tract IgA antibodies to different hemagglutinin molecules. Eur J Immunol. 1991;21:1337–44. DOIPubMedGoogle Scholar
- Davidson GP, Whyte PB, Daniels E, Franklin K, Nunan H, McCloud PI, Passive immunisation of children with bovine colostrum containing antibodies to human rotavirus [see comments]. Lancet. 1989;2:709–12. DOIPubMedGoogle Scholar
- Ebina T. Prophylaxis of rotavirus gastroenteritis using immunoglobulin. Arch Virol Suppl. 1996;12:217–23.PubMedGoogle Scholar
- Weltzin R, Traina-Dorge V, Soike K, Zhang JY, Mack P, Soman G, Intranasal monoclonal IgA antibody to respiratory syncytial virus protects rhesus monkeys against upper and lower respiratory tract infection. J Infect Dis. 1996;174:256–61.PubMedGoogle Scholar
- Peterson EM, Cheng X, Motin VL, de la Maza LM. Effect of immunoglobulin G isotype on the infectivity of Chlamydia trachomatis in a mouse model of intravaginal infection. Infect Immun. 1997;65:2693–9.PubMedGoogle Scholar
- Lyerly DM, Bostwick EF, Binion SB, Wilkins TD. Passive immunization of hamsters against disease caused by Clostridium difficile by use of bovine immunoglobulin G concentrate. Infect Immun. 1991;59:2215–8.PubMedGoogle Scholar
- Tacket CO, Losonsky G, Link H, Hoang Y, Guesry P, Hilpert H, Protection by milk immunoglobulin concentrate against oral challenge with enterotoxigenic Escherichia coli. N Engl J Med. 1988;318:1240–3.PubMedGoogle Scholar
- Booth V, Ashley F, Lehner T. Passive immunization with monoclonal antibodies against Porphyromonas gingivalis in patients with periodontitis. Infect Immun. 1996;64:422–7.PubMedGoogle Scholar
- Tacket C, Binion S, Bostwick E, Losonsky G, Roy M, Edelman R. Efficacy of bovine milk immunoglobulin concentrate in preventing illness after Shigella flexneri challenge. Am J Trop Med Hyg. 1992;47:276–83.PubMedGoogle Scholar
- Ramisse F, Szatanik M, Binder P, Alonso J-M. Passive local immunotherapy of experimental staphylococcal pneumonia with human intravenous immunoglobulin. J Infect Dis. 1993;168:1030–3.PubMedGoogle Scholar
- Ma JK, Hunjan M, Smith R, Kelly C, Lehner T. An investigation into the mechanism of protection by local passive immunization with monoclonal antibodies against Streptococcus mutans. Infect Immun. 1990;58:3407–14.PubMedGoogle Scholar
- Apter FM, Michetti P, Winner LSD, Mack JA, Mekalanos JJ, Neutra MR. Analysis of the roles of antilipopolysaccharide and anti-cholera toxin immunoglobulin A (IgA) antibodies in protection against Vibrio cholerae and cholera toxin by use of monoclonal IgA antibodies in vivo. Infect Immun. 1993;61:5279–85.PubMedGoogle Scholar
- Cassone A, Boccanera M, Adriani D, Santoni G, De Bernardis F. Rats clearing a vaginal infection by Candida albicans acquire specific, antibody-mediated resistance to vaginal reinfection. Infect Immun. 1995;63:2619–24.PubMedGoogle Scholar
- Perryman LE, Riggs MW, Mason PH, Fayer R. Kinetics of Crytosporidium parvum sporozoite neutralization by monoclonal antibodies, immune bovine serum, and immune bovine colostrum. Infect Immun. 1990;58:257–9.PubMedGoogle Scholar
- Ma JK, Hein MB. Immunotherapeutic potential of antibodies produced in plants. Trends Biotechnol. 1995;13:522–7. DOIPubMedGoogle Scholar
- Chintalacharuvu KR, Morrison SL. Production of secretory immunoglobulin A by a single mammalian cell. Proc Natl Acad Sci U S A. 1997;94:6364–8. DOIPubMedGoogle Scholar
- Kilian M, Russel M. Function of mucosal immunoglobulins. In: Ogra P, Mestecky J, Lamm M, Strober W, McGhee J, Bienenstock J, editors. Handbook of mucosal immunology. San Diego: Academic Press; 1994. p. 127-37.
- Kraehenbuhl JP, Neutra MR. Molecular and cellular basis of immune protection of mucosal surfaces. Physiol Rev. 1992;72:853–79.PubMedGoogle Scholar
- Winter G, Griffiths A, Hawkins R, Hoogenboom H. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12:433–55. DOIPubMedGoogle Scholar
- Burton D, Barbas C. Human antibodies from combinatorial libraries. Adv Immunol. 1994;57:191–280. DOIPubMedGoogle Scholar
- Williamson R, Burioni R, Sanna P, Partridge L, Barbas C, Burton D. Human monoclonal antibodies against a plethora of viral pathogens from single combinatorial libraries. Proc Natl Acad Sci U S A. 1993;90:4141–5. DOIPubMedGoogle Scholar
- Vaughan T, Williams A, Pritchard K, Osbourn J, Pope A, Earnshaw J, Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–14. DOIPubMedGoogle Scholar
- Green LL, Hardy MC, Maynard-Currie CE, Tsuda H, Louie DM, Mendez MJ, Antigen-specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nat Genet. 1994;7:13–21. DOIPubMedGoogle Scholar
- Mendez MJ, Green LL, Corvalan JR, Jia XC, Maynard-Currie CE, Yang XD, Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat Genet. 1997;15:146–56. DOIPubMedGoogle Scholar
- Sherman-Gold R. Monoclonal antibodies: the evolution from '80s magic bullets to mature, mainstream applications as clinical therapeutics. Genetic Engineering News 1997:17.
- Vaughan TJ, Osbourn JK, Tempest PR. Human antibodies by design. Nat Biotechnol. 1998;16:535–9. DOIPubMedGoogle Scholar
- Barbas C, Hu D, Dunlop N, Sawyer L, Cababa D, Hendry R, In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc Natl Acad Sci U S A. 1994;91:3809–13. DOIPubMedGoogle Scholar
- Barbas CF, Burton DR. Selection and evolution of high-affinity human anti-viral antibodies. Trends Biotechnol. 1996;14:230–4. DOIPubMedGoogle Scholar
- Crothers D, Metzger H. The influence of polyvalency on the binding properties of antibodies. Immunochemistry. 1972;9:341–57. DOIPubMedGoogle Scholar
- Sherwood JK, Zeitlin L, Chen X, Whaley KJ, Cone RA, Saltzman WM. Residence half-life of IgG administered topically to the mouse vagina. Biol Reprod. 1996;54:264–9. DOIPubMedGoogle Scholar
- Glassy M. Production: the rate-limiting step in obtaining human monoclonal antibody pharmaceuticals. In: Monoclonal antibody production. La Jolla (CA): International Business Communications; 1996.
- DeYoung G. Monoclonal Ab processors/manufacturers stress costs and productivity. Genetic Engineering News 1996:8.
- Hiatt A, Caffertey R, Bowdish K. Production of antibodies in transgenic plants. Nature. 1989;342:76–8. DOIPubMedGoogle Scholar
- Genzyme transgenic manufactures monoclonal antibody in goats' milk [press release]. Cambridge (MA): Genzyme; May, 1995.
- Seaver S. Monoclonal antibodies: using new techniques to reduce development time. Genetic Engineering News 1997:13.
- Food and Drug Administration. International conference on harmonisation; guidance on preclinical safety evaluation of biotechnology-derived pharmaceuticals; availablity. Fed Regist. 1997;62:61515–9.
- OraVax Reports Results from Phase III Trial of HNK20 Nosedrop for Respiratory Syncytial Virus in Infants [press release]. Cambridge (MA): OraVax; March 19, 1997.
- Ma J, Smith R, Lehner T. Use of monoclonal antibodies in local passive immunization to prevent colonization of human teeth by Streptococcus mutans. Infect Immun. 1987;55:1274–8.PubMedGoogle Scholar
- Beck L, Boots L, Stevens V. Absorption of antibodies from the baboon vagina. Biol Reprod. 1975;13:10–6. DOIPubMedGoogle Scholar
- Corthesy B, Kaufmann M, Phalipon A, Peitsch M, Neutra M, Kraehenbuhl J-P. A pathogen-specific epitope inserted into recombinant secretory immunoglobulin A is immunogenic by the oral route. J Biol Chem. 1996;271:33670–7. DOIPubMedGoogle Scholar
- Tsume Y, Taki Y, Sakane T, Nadai T, Sezaki H, Watabe K, Quantitative evaluation of the gastrointestinal absorption of protein into the blood and lymph circulation. Biol Pharm Bull. 1996;19:1332–7.PubMedGoogle Scholar
- Kuo PY, Sherwood JK, Saltzman WM. Topical antibody delivery systems produce sustained levels in mucosal tissue and blood. Nat Biotechnol. 1998;16:163–7. DOIPubMedGoogle Scholar
- Points to consider in the manufacture and testing of monoclonal antibody products for human use. Washington: U.S. Department of Health and Human Services, Food and Drug Administration; 1997.
- Ma JK, Hein MB. Plant antibodies for immunotherapy. Plant Physiol. 1995;109:341–6. DOIPubMedGoogle Scholar
- Ma JK, Hikmat BY, Wycoff K, Vine ND, Chargelegue D, Yu L, Characterization of a recombinant plant monoclonal secretory antibody and preventive immunotherapy in humans. Nat Med. 1998;4:601–6. DOIPubMedGoogle Scholar
- Mo H, Stamatatos L, Ip J, Barbas C, Parren P, Burton D, Human immunodeficiency virus type 1 mutants that escape neutralization by human monoclonal antibody IgG1b12. J Virol. 1997;71:6869–74.PubMedGoogle Scholar
- Babcook JS, Leslie KB, Olsen OA, Salmon RA, Schrader JW. A novel strategy for generating monoclonal antibodies from single, isolated lymphocytes producing antibodies of defined specificities. Proc Natl Acad Sci U S A. 1996;93:7843–8. DOIPubMedGoogle Scholar
- Murray C, Lopez A. Global and regional cause-of-death patterns in 1990. In: Murray C, Lopez A, editors. Global comparative assessments in the health sector. Geneva: World Health Organization; 1994. p. 21-54.
- Eng T, Butler W. The hidden epidemic. Institute of Medicine. Washington: National Academy Press; 1997.
- Bogstedt A, Johansen K, Hatta H, Kim M, Casswall T, Svensson L, Passive immunity against diarrhoea. Acta Paediatr. 1996;85:125–8. DOIPubMedGoogle Scholar
- Eibl M, Wolf H, Furnkranz H, Rosenkranz A. Prevention of necrotizing enterocolitis in low-birth-weight infants by IgA-IgA feeding. N Engl J Med. 1988;319:1–7.PubMedGoogle Scholar
- Petschow B, Talbott R. Reduction in virus-neutralizing activity of a bovine colostrum immunoglobulin concentrate by gastric acid and digestive enzymes. J Pediatr Gastroenterol Nutr. 1994;19:228–35. DOIPubMedGoogle Scholar
- Ogra P, Fishaut M. Human breast milk. In: Remington J, Klein J, editors. Infectious diseases of the fetus and newborn infant. 3rd ed. Philadelphia: W.B. Saunders Company; 1990. p. 68-84.
- Prince G, Hemming V, Horswood R, Baron P, Chanock R. Effectiveness of topically administered neutralizing antibodies in experimental immunotherapy of respiratory syncytial virus infection in cotton rats. J Virol. 1987;61:1851–4.PubMedGoogle Scholar
- Radomsky ML, Whaley KJ, Cone RA, Saltzman WM. Controlled vaginal delivery of antibodies in the mouse. Biol Reprod. 1992;47:133–40. DOIPubMedGoogle Scholar
- Sherwood J, Zeitlin L, Whaley K, Cone R, Saltzman W. Controlled release of antibodies for long-term topical passive immunoprotection of female mice against genital herpes. Nat Biotechnol. 1996;14:468–71. DOIPubMedGoogle Scholar
- Tjokronegoro A, Sirisinha S. Degradation of immunoglobulins by secretions of human reproductive tracts. J Reprod Fertil. 1974;38:221–4. DOIPubMedGoogle Scholar
- Singh M, Sugathan PS, Bhujwala RA. Human colostrum for prophylaxis against sticky eyes and conjunctivitis in the newborn. J Trop Pediatr. 1982;28:35–7.PubMedGoogle Scholar
Figures
Tables
Cite This ArticleTable of Contents – Volume 5, Number 1—February 1999
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Kevin Whaley, 3400 North Charles St. Jenkins Hall, Baltimore, MD 21218, USA; fax: 410-516-6597
Top