Volume 27, Number 4—April 2021
Research
Evolution of Sequence Type 4821 Clonal Complex Hyperinvasive and Quinolone-Resistant Meningococci
Abstract
Expansion of quinolone-resistant Neisseria meningitidis clone ChinaCC4821-R1-C/B from sequence type (ST) 4821 clonal complex (CC4821) caused a serogroup shift from serogroup A to serogroup C invasive meningococcal disease (IMD) in China. To determine the relationship among globally distributed CC4821 meningococci, we analyzed whole-genome sequence data from 173 CC4821 meningococci isolated from 4 continents during 1972–2019. These meningococci clustered into 4 sublineages (1–4); sublineage 1 primarily comprised of IMD isolates (41/50, 82%). Most isolates from outside China (40/49, 81.6%) formed a distinct sublineage, the Europe–USA cluster, with the typical strain designation B:P1.17-6,23:F3-36:ST-3200(CC4821), harboring mutations in penicillin-binding protein 2. These data show that the quinolone-resistant clone ChinaCC4821-R1-C/B has expanded to other countries. The increasing distribution worldwide of serogroup B CC4821 raises the concern that CC4821 has the potential to cause a pandemic that would be challenging to control, despite indirect evidence that the Trumenba vaccine might afford some protection.
Neisseria meningitidis, a leading cause of bacterial meningitis and septicemia globally, causes ≈1.2 million invasive meningococcal disease (IMD) cases annually and a case-fatality rate of 11% (1). Meningococci are classified into 12 serogroups based on capsular polysaccharides (1); genetic relationships among isolates are defined by clonal complexes (CCs) identified by multilocus sequence typing (MLST), which are surrogates for lineages (2). The relationship among serogroups, CCs (lineages), and IMD fluctuates over time and by location, but IMD isolates are dominated by CCs known as hyperinvasive lineages, usually associated with one of the 6 disease-causing serogroups (MenA, MenB, MenC, MenW, MenX, and MenY).
In China, the national dissemination of hyperinvasive sequence type (ST) 4821 clonal complex (CC4821) meningococci led to a shift in IMD epidemiology from mostly MenA to predominantly MenC (3,4). Although no quinolone resistance was identified in CC4821 in China during 1965–1985, high-frequency resistance (79%) occurred from 2005 onward due to expansion of the quinolone-resistant clone ChinaCC4821-R1-C/B (5). Previous studies discovered that CC4821 can be divided into 2 groups, with group 1 associated with IMD (6,7). Peng et al. identified 6 strain-specific genome regions resulting from horizontal gene transfer (HGT) in isolate 053442 (8); this finding was consistent with the emergence of the ChinaCC4821-R1-C/B clone associated with multiple HGT events within genes encoding surface antigens (6), although the donors of these events were not identified.
Figure 1
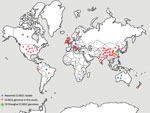
Figure 1. Distribution of CC4821 Neisseria meningitidisisolates worldwide. CC4821 isolates were identified in China and in 19 countries of Europe, Africa, North America, South America, Oceania, and Asia. CC, clonal...
Globally, the number of CC4821 IMD isolates has increased. At the time CC4821 was identified, isolates were confined to China (4,9); however, by June 2020, a total of 59 CC4821 isolates had been identified in 19 countries worldwide (Figure 1). Moreover, 3 IMD cases caused by quinolone-resistant CC4821 isolates were reported in Canada (n = 2) and Japan (n = 1) after 2013 (10,11); 3 other CC4821 isolates were found to colonize the anorectal tract of men who have sex with men (MSM) (12). We investigated the genomic events leading to the emergence and expansion of hyperinvasive CC4821 meningococci by describing the phylogenetic relationships among meningococci with different serogroups (MenC, MenB, MenW, and nongroupable), sources (IMD, carriage, and MSM), locations (China or other countries), and dates of isolation (1972–1978 vs. 2004–2019). We assessed genes encoding key antigens and antimicrobial resistance phenotypes, identified putative donors of HGT events unique to the epidemic and quinolone-resistant clone ChinaCC4821-R1-C/B, and characterized isolates outside of China.
Isolate Collection and Whole-Genome Sequencing
A total of 173 CC4821 genomes were collected dating from 1972–1978 (n = 19) and 2004–2019 (n = 154), including isolates from IMD (66/173, 38.2%), genitourinary sites (6/173, 3.5%), asymptomatic carriage (86/173, 49.7%), and unknown sources (15/173, 8.7%) (Appendix 1 Table 1). Shanghai CDC sequenced 76 CC4821 isolates with Illumina HiSeq (Illumina, https://www.illumina.com) using paired-end 150 base reads as previously described (13). An additional 97 publicly available CC4821 genomes consisted of 48 genomes from 14 provinces of China, including the reference strain 053442 (6–8) and 49 genomes from countries outside of China, including the United Kingdom (n = 20), United States (n = 8), and 11 other countries (n = 21) (Figure 1; Appendix 1 Table 1) (10,12,14–17). The completeness and contamination of the genomes were evaluated using CheckM (18).
Antigenic and Antimicrobial Resistance Characteristics of CC4821 Genomes
To describe the antigenic and antimicrobial resistance characteristics of CC4821 genomes, we extracted from genomes nucleotides of 9 antigen coding genes (porA, fHbp, nhba, porB, fetA, opcA, nspA, tbpA, and NMB0315) (19–22) and 5 resistance-associated genes (gyrA, parC, penA, ponA, and rpoB) (23,24) for analysis. We annotated and analyzed deduced encoding factor H–binding protein (fHbp), Neisseria heparin-binding antigen (NHBA), Neisseria adhesion antigen (NadA), and outer membrane protein (PorA) peptides and deduced meningococcal vaccine antigen reactivity (MenDeVAR) index from the PubMLST Neisseria database (25).
Identifying CC4821 (L44) Sublineages
In the Neisseria PubMLST database, a lineage-specific core genome MLST typing scheme containing loci found in 95% of CC4821 isolates was established and designated L44 cgMLST consistent with the previously described CC4821 lineage 44 (26). We compared the 173 CC4821 genomes using Genome Comparator (27) and the L44 cgMLST scheme, identifying distinct sublineages. To characterize each sublineage, we visualized a FASTA output from the Genome Comparator Tool using all 2,860 defined loci (NEIS0001–NEIS3173, not contiguous) using MEGA version 5 (28). We used Z2491 (GenBank accession no. NC_003116) as outgroup in accordance with previous studies (6,8). Assembled contigs and annotation information of 173 genomes in this study can be accessed at https://pubMLST.org/neisseria (Appendix 1 Table 1).
Identifying and Characterizing Unique Alleles in Sublineages
We determined shared and unique alleles using outputs from Genome Comparator. An allele was defined as unique to a sublineage if it was present in >90% of the genomes in that sublineage but absent in other sublineages. Genes with unique alleles were functionally characterized according to the Kyoto Encyclopedia of Genes and Genomes Orthology groupings of its database (29).
Identifying HGT Events and Putative Donors
Inputting the aligned sequences generated from Parsnp (30), we predicted putative HGT events using Gubbins (31). To search for potential donors, we blasted alleles and sequences of contiguous loci that were predicted to originate from HGT against the PubMLST database. We identified potential donors as previously described (32). We labeled recombination areas with unique loci on the circular genome map of genome 053442 by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) comparisons to strains of other sublineages, as generated using BRIG (33).
Screening Molecular Markers of MSM Infection Strains from Europe
In addition to the lineage of 11.2 possessing PorA P1.5–1,10–8, 3 other molecular features have been identified in meningococci causing infections among MSM in Europe during 2012–2014; these features were functional nitrite reductase (AniA); frameshifted fHbp allele found mostly in urethritis and proctitis isolates; and penA327 that had reduced susceptibility to penicillin and third-generation cephalosporins (34). These 3 molecular markers were screened among all the 173 CC4821 genomes.
Isolate Characterization
The 173 CC4821 isolates represented 46 different STs; ST4821 (n = 41, 23.7%) and ST3200 (n = 30, 17.3%) were the most prevalent. We identified 43 PorA subtypes, of which P1.7-2,14 (n = 25, 14.5%) and P1.17-6,23 (n = 18, 10.4%) were the most frequent. We identified 27 FetA variants; F3-3 (n = 47, 27.2%) and F3-36 (n = 37, 21.4%) were the most prevalent (Appendix 1 Table 1).
Identifying 4 Sublineages
Figure 2
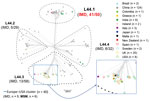
Figure 2. Allele-based sublineages of clonal complex 4821 Neisseria meningitidisidentified using lineage 44 core genome multilocus sequence typing scheme. The inset shows the country distribution of the 40 genomes constituting...
Figure 3

Figure 3. Phylogenetic tree and data of clonal complex 4821 Neisseria meningitidis sublineage L44.1 (ChinaCC4821-R1-C/B) isolates. Red text indicates the oldest isolate of the sublineage; blue text, the...
We identified 2,161 loci in reference genome 053442, including 1,699 core genes. Most (1,527/1,699, 89.9%) of the core loci had p-distance values of 0–0.1; 0.8% (14/1,699) showed high p-distance values of 0.50–0.68. On the basis of the L44 cgMLST scheme, we divided the CC4821 isolates into 4 sublineages (Figure 2): L44.1, identical to the ChinaCC4821-R1-C/B clone (n = 50, 28.9%), composed of isolates from China (n = 44) and other countries (n = 6) during 2004–2019 that were very closely related (Figure 3); L44.2 (n = 29, 16.8%), composed of isolates from China (n = 28) and the United Kingdom (n = 1) during 2005–2019; L44.3 (n = 58, 33.5%), composed of isolates from China (n = 18) and countries outside China (n = 40) during 1977–2019; and L44.4 (n = 32, 18.5%), composed of isolates from China (n = 30) and India (n = 2) during 1972–2017. Four additional isolates from China were not assigned to any sublineages.
Features of the 4 Sublineages
The percentage of IMD isolates was significantly higher in L44.1 (41/50, 82%) than the other 3 sublineages (17.2%–22.4%; p<0.001) (Figure 2). L44.1, containing the reference strain 053442, was mainly composed of MenC isolates (44/50, 88%) and had ST4821 as its central ST. L44.2, was mainly composed of MenB isolates (27/29, 93.1%) and its central ST was ST5664. L44.3 was mainly composed of MenB (55/58, 94.8%) with ST3200 as its central ST. L44.4 was mainly composed of MenC (14/32, 43.8%) and MenW (11/32, 34.4%) with its central ST3436 (Appendix 2 Figure 1).
Figure 4

Figure 4. Genomic diversity of clonal complex 4821 Neisseria meningitidis sublineage L44.1 (ChinaCC4821-R1-C/B) isolates. The numbers underneath the antigen genes and AMR genes are the dominant alleles for...
Figure 5

Figure 5. Phylogenetic tree and data of clonal complex 4821 Neisseria meningitidissublineage L44.3 isolates. Red text indicates the oldest isolates of the sublineage; blue text, the isolates from countries outside...
Figure 6

Figure 6. Genomic diversity of clonal complex 4821 Neisseria meningitidissublineage L44.3 isolates. The numbers underneath the antigen genes and AMR genes are the dominant alleles for that particular gene, and...
Analysis of the 5 antimicrobial resistance genes revealed that both gyrA-71 (with T91I) and parC-12 were specific to L44.1; parC-275 and penA-9 (with 5 mutations) were both specific to L44.3, and gyrA-294 (with T91I) was discovered only in L44.4 (Table 1; Appendix 2 Figures 2–6). In L44.1, all of the isolates possessed the quinolone resistance–associated mutation T91I in GyrA (Figure 4). In L44.3, 40/58 (69.0%) harbored PBP2 mutations, almost always from countries outside of China (38/40, 95%) (Figures 5, 6).
Vaccine Antigens among the 4 Sublineages
Analysis of 9 antigenic genes identified several alleles unique to a certain sublineage (Table 2; Appendix 2 Figures 7–17). For example, FetA-VR F3-3 was found in L44.1, F1-91 in L44.2, F3-36 and F3-9 in L44.3, and F1-7 in L44.4 isolates (Appendix 2 Figure 11). In L44.1, most isolates had the same antigenic gene profile (nhba-124, porB-29, fetA-64, opcA-4, nspA-4, tbpA-7, and NMB0315-21) (Figure 4), and 25/50 (50%) had the PorA subtype of P1.7-2,14 (Figure 3). In L44.3, most had the same gene profile (fHbp-16, nhba-553, porB-265, fetA-1069, opcA-100, nspA-26, and NMB0315-194), with porA and tbpA showing high genetic diversity (Figure 5).
We analyzed deduced peptide sequences for vaccine antigen constituents among MenB isolates (n = 97). We identified 16 fHbp peptides, of which peptide 16 (variant 2/subfamily A) was present in 70/97 (72.2%) isolates, including 31/70 isolates from China. There were 20 NHBA peptides, of which alleles 669 (46/95, 48.4%), 901 (11/95, 11.6%), and 668 (10/95, 10.5%) occurred most frequently. The nadA gene was absent in all isolates (including other serogroups). Of 31 PorA VR1/VR2 combinations, the most frequently occurring was P1.20,23 (11/97, 11.3%).
MenDeVAR Index values were assigned for MenB disease isolates (n = 29, including the 6 isolates from genitourinary sites), but 27/29 (93.1%) isolates had insufficient data from experimental studies to estimate the coverage of the MenB vaccine Bexsero (Appendix 2 Table 2). We predicted cross-reactivity to the MenB vaccine Trumenba for 18/29 (62.1%) isolates. For the MenB disease isolates from China, 7/17 (41.2%) were deemed cross-reactive with Trumenba; however, we had insufficient data for the remaining 10/17 (58.8%) to determine reactivity.
Molecular Markers of Strains from Europe Infecting MSM
None of the CC4821 isolates harbored frameshifted fHbp allele or penA327, but the distribution of putatively functional AniA proteins was diverse. The aniA gene was absent in all L44.1 isolates (Figure 3) but was present in all of the other 123 CC4821 isolates, of which 96.7% (119/123) isolates harbored putatively functional AniA proteins (Figure 5; Appendix 2 Figures 16–17).
Evolution of Sublineage L44.1 (ChinaCC4821-R1-C/B Clone)
Five specific loci were present in >90% of L44.1 but in <10% of other sublineages. These loci were involved in signaling and cellular processes (n = 2), metabolism (n = 1), and genetic information processing (n = 1) (Table 3). No loci were specific to any of other 3 sublineages.
Figure 7

Figure 7. Circular genome map of CC4821 Neisseria meningitidis genome 053442 with BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) comparisons to the genomes of other sublineages. The innermost rings show guanine and cytosine...
Prediction of HGT events contributing to the emergence of L44.1 using Gubbins discovered 126 events involving 686 loci shared by the 50 L44.1 isolates (Appendix 2 Figure 18). These events included 216 loci with alleles specific to L44.1. We discovered an additional 83 unique loci based on analysis of the accessory genome. Therefore, a total of 299 unique loci were identified in L44.1; of those, 139 (46.5%) were involved in metabolic function (Appendix 1 Table 3). These 299 unique loci were distributed across the chromosome; we observed 44 areas (216 loci) harboring contiguous loci with unique alleles (Figure 7), among which the exact donors of 36 areas across 149 loci were identified in 46 putative HGT events. The total length of these putative recombination fragments was ≈225 kb, including 87 kb (38.7%) originating from the C:ST-9514 cluster isolates in China during 1966–1977, followed by 25 kb (11.1%) from MenA isolates (CC5 and CC1) in China during 1966–1984 (Table 4).
Evolution of CC4821 Isolates from Outside China
We identified 49 CC4821 isolates from countries outside of China, and most (40/49, 81.6%) were assigned to L44.3, of which there were 39 MenB and 1 MenC, constituting the distinct Europe–USA cluster (Figures 2, 5). The representative molecular characteristics of the Europe–USA cluster was B:P1.17-6,23: F3-36:ST-3200(CC4821); its antigen gene profile was porA-423, fHbp-16, nhba-553, porB-265, fetA-1069, opcA-100, nspA-26, tbpA-1333, and NMB0315-194 and antimicrobial resistance profile gyrA-12, parC-275, penA-9 with PBP2 mutations, ponA-7, and rpoB-85 (Figure 6). In Gubbins analysis, 33 events involving 193 loci were shared by all the Europe–USA cluster isolates (Appendix 2 Figure 18); we discovered 60 unique loci for which we could not identify their potential donors. These unique loci were involved in functions mainly associated with metabolism (23/60, 38.3%) and genetic information processing (18/60, 30%) (Appendix 1 Table 4).
In addition to the 40 Europe–USA cluster isolates, there were 6 MenC invasive isolates from India (n = 4, identified 2014–2016), Japan (n = 1, identified in 2017), and New Zealand (n = 1, identified in 2018). These 6 isolates were clustered together and were closely related with 44 isolates from China within sublineage L44.1 (Figures 2–3). Only the isolate from Japan showed the typical molecular feature of Anhui outbreak strain (C:P1.7-2,14:F3-3:ST-4821[CC4821]).
Features and Evolution of Serogroup W CC4821 Isolates
A total of 11 MenW isolates from China were identified; the representative strain designation was W:P1.5-3,10-2:F1-7:ST-8491(CC4821), with similar gene profiles of antigen-encoding loci (porA-1804, fHbp-474, nhba-966, fetA-37, opcA-4, nspA-117, and NMB0315-21) and antimicrobial resistance loci (gyrA-294 with T91I, parC-779, ponA-7, and rpoB-85). These MenW isolates constituted a distinct cluster in L44.4; they were more closely related to NM193 (C:P1.20-3,23-1:F1-5:ST-3436[CC4821], dating from 1972) than to NM205 (C:P1.20,23-2:F5-135:ST-4821[CC4821], dating from 1973) (Appendix 2 Figure 17).
The meningococci can cause IMD, leading to endemic disease in most if not all human populations. Several genotypes belonging to hyperinvasive lineages, in combination with the disease-associated capsular serogroups, can cause elevated levels of disease; some of which also possess epidemic and pandemic potential. In the past 100 years, notable epidemics and pandemics have included meningococci such as A:CC1, A:CC5, B:CC41/44, C:CC11, and W:CC11 (35). Here, we employed a genomic analysis of MenB, MenC, and MenW CC4821 isolates dating from 1972–2019 to assess their epidemic and pandemic potential. Of special concern are the expansion of the quinolone-resistant clone ChinaCC4821-R1-C/B from China to other countries; the potential possession of universal resistance to penicillin in Europe–USA cluster isolates; and the uncertainty over the potential efficacy of existing vaccines to prevent B:CC4821 diseases.
CC4821, which corresponds to lineage 44, shares several properties in common with the hyperinvasive CC11 meningococci (Lineage 11): its ability to express several serogroups, global distribution, colonization of urogenital and anorectal tracts, and separation into distinct sublineages. CC11 has caused well-documented epidemics and pandemics on several occasions, including US military outbreaks in the 1960s; Hajj-associated outbreaks in 2000s; and the global epidemics from 2010, especially outbreaks among MSM (34–38). These similar characteristics raise the concern that the CC4821 may have the potential to cause similar global pandemics.
Consistent with the presence of the epidemic CC4821 clone in countries outside of China, 6 CC4821 IMD meningococci from India, Japan, and New Zealand, isolated during 2014–2018, clustered with ChinaCC4821-R1-C/B meningococci in L44.1 (Figure 3). IMD cases caused by these 6 isolates were all found in native inhabitants (10,15,39); all 6 isolates shared similar serogroup, antibiotic, and antigen (except porA and fHbp) gene characteristics with isolates from China in this sublineage. In particular, all 6 isolates harbored the T91I mutation in GyrA, the molecular marker of quinolone resistance, compatible with their quinolone-resistant phenotype (10,15,39). These isolates also had strain-specific features, which suggested that they resulted from transmission from the ChinaCC4821-R1-C/B clone. The isolate from Japan, which had the typical molecular features of the Anhui outbreak strain (C:P1.7-2,14:F3-3:ST-4821[CC4821]) (4), became the earliest-reported quinolone-resistant meningococcus harboring ParC mutation (S87I, allele 1538) to cause IMD worldwide (10). In contrast, none of the ChinaCC4821-R1-C/B clone isolates from China had ParC mutations conferring antimicrobial resistance. The isolate from Japan was more closely related to the reference strain 053442 than were the 4 India and 1 New Zealand isolates, which had different STs, porA, fHbp, and tbpA alleles (Figure 4).
Although we did not identify a putative ancestor of the quinolone-resistant clone ChinaCC4821-R1-C/B in this study, we found 299 loci with alleles unique to this sublineage. Approximately half of these loci were associated with metabolic pathways, suggesting that divergence in metabolic genes may play a role in the emergence of epidemic meningococci. Several studies have indicated that metabolic genes can influence the pathogenesis and virulence of the meningococcus, for example by allowing alternative host resources to be exploited in invasive disseminated infections (40). Changes in the hyperinvasive A:CC5 meningococci circulating in Africa have been associated with HGT of core genes involved in metabolic processes (41). The putative donors of these unique alleles included lineages from different serogroups and dates of isolation, such as C:ST-9514 cluster, 1960s–1970s; A:CC5 and A:CC1, 1960s–1980s; B:CC32, 1960s; B:CC41/44, 1970s; and E:CC178, 1980s (Table 4). The C:ST-9514 cluster, STs that do not presently form part of a clonal complex documented in PubMLST, has ST9514 as the central ST and was predominant in MenC carriage isolates during 1965–1980 in Shanghai, China (42). Therefore, the emergence of ChinaCC4821-R1-C/B clone was perhaps associated with accumulation of these unique alleles, which accounted for the separation from other sublineages in the allele-based phylogeny (Figure 2).
In the PubMLST database, >60% of the CC4821 isolates from outside China were MenB. Of these, 49 genomes were available in this study, including isolates from IMD (n = 15) and urogenital and rectal tracts (n = 6). Most of these genomes clustered in sublineage L44.3 and constituted a distinct cluster, the Europe–USA cluster, showing the typical strain designation: B:P1.17-6,23-x:F3-36:ST-3200(CC4821), wherein 23-x refers to 23, 23-2, and 23-6. The PorA and FetA types P1.17-6,23-x and F3-36 were only found in this cluster. The Neisseria PubMLST database had no genome data for 24 CC4821 isolates from other countries (United States, Brazil, France, Czech Republic, Spain, Italy, Australia, and Vietnam), but included PorA or FetA variants for the 24 isolates (Appendix 1 Table 5). Of these, 19 (79.2%) exhibited P1.17-6,23-x or F3-36, suggesting they might belong to the Europe–USA cluster. This cluster was distinct from the epidemic clone ChinaCC4821-R1-C/B. For example, the antigen profile characteristic of the Europe–USA cluster was P1.17-6,23-x, F3-36, PorB-3-229, fHbp-16, nhba-553, opcA-100, nspA-26, and tbpA-1333, compared with P1.7-2,14, F3-3, PorB-3-48, fHbp-498, 22 and 489, nhba-124, opcA-4, nspA-4, and tbpA-7 in the ChinaCC4821-R1-C/B clone. In addition, all the ChinaCC4821-R1-C/B isolates harbored the mutation T91I in GyrA, whereas almost all of the Europe–USA cluster isolates possessed mutations in PBP2 (F504L, A510V, I515V, H541N, and I566V). This may reflect different antibiotic selective pressures experienced by the Europe–USA and the ChinaCC4821-R1-C/B meningococci. Penicillins were the most-used antimicrobial drugs in outpatients in Europe, whereas China has the second largest global increases of fluoroquinolone consumption (43,44). A high frequency (>70%) of quinolone resistance has been reported in China since 2005 (5), whereas 65% of meningococci in Europe showed reduced susceptibility to penicillin G during 1945–2006 (45). In the 2 oldest isolates of the sublineage L44.3, Nm282 (B:P1.20,23:F3-36:ST-3200[CC4812]) was much closer to the Europe–USA cluster isolates than Nm323 (B:P1.20,23:F3-36:ST-5798[CC4821]) (Figure 5), and it seemed more likely to be the ancestor of the Europe–USA cluster isolates.
Urogenital and rectal meningococci have raised increasing public health concerns (34). In 2017, CC4821 anorectal isolates were identified in the United Kingdom (12). In this study, we identified CC4821 isolates from urethral and rectal tracts that clustered with isolates from IMD specimens and oropharyngeal carriage (Figure 5). With the exception of L44.1 isolates, most of the CC4821 isolates contained a putatively functional nitrite reductase (AniA), required for growth in anaerobic environments. The CC4821 isolates acquired quinolone resistance alleles from N. lactamica and N. subflava (46); the ability to grow in anaerobic environments will facilitate acquisition of gonococcal alleles, including antimicrobial resistance alleles. Such events seem to have already occurred in a sublineage of CC11, which was responsible for several IMD outbreaks and urethritis among MSM (34). They shared the same penA allele (penA327/penAXXXIV) with gonococcal bacteria and showed decreased susceptibility to third-generation cephalosporins (47). Although PubMLST is the largest global repository of meningococcal genomes (>22,000), a paucity of genomic data were available from isolates originating from the genitourinary or respiratory tract, suggesting an underestimation of the global dissemination of CC4821. Therefore, we recommend WGS for urogenital-, rectal-, and respiratory-derived meningococci if they are exhibiting antimicrobial resistance.
CC4821 lineage 44 includes isolates from different serogroups, including MenB, MenC, and MenW. In China, MenC and MenW isolates can be prevented by vaccines, such as group A and C meningococcal polysaccharide vaccine (MPV-AC) and MPV-ACYW, but no routinely administered vaccine is available to prevent MenB IMD (48). Two protein-based vaccines targeting MenB meningococci, 4CMenB (Bexsero) and rLP2086 (Trumenba), have been licensed in several countries (49–50; reference 51 in Appendix 2), but limited data are available on the bacterial coverage of these vaccines to CC4821 isolates directly from serum bactericidal activity assays, the Meningococcal Antigen Typing System (MATS) for Bexsero, or meningococcal antigen surface expression for Trumenba. One B:CC4821 isolate (M14-240580, UK) was reported to be tested using the MATS assay and showed no potential protection (reference 52 in Appendix 2). Using systems to index complex genotypic and phenotypic data, such as the MenDeVAR Index, we predicted that ≈60% of B:CC4821 disease-causing isolates might be prevented through vaccination with Trumenba; data are insufficient to infer Bexsero reactivity. Further testing of globally diverse meningococci is needed with these experimental assays to analyze potential vaccine impact in settings outside Europe.
In summary, we have undertaken a comprehensive genomic analysis of a hyperinvasive meningococcal CC4821 expressing MenB, MenC, and MenW with expansion from China to other global geographic locations with currently available genomic data. We identified key genomic factors and putative evolutionary changes that might have led to the emergence and persistence of the epidemic quinolone-resistant clone in China. Vaccine coverage for MenB CC4821 isolates needs further evaluation. Enhanced laboratory surveillance for CC4821 isolates from IMD cases and from oropharyngeal, urethral, and rectal carriage is needed to monitor global trends of expansion, which will be essential for local immunization policies.
Dr. Chen is an associate professor in Department of Microbiology, Shanghai Municipal Center for Disease Control and Prevention. His research interests include mechanisms of antimicrobial resistance in clinical isolates responsible for respiratory tract infections.
Acknowledgments
We thank the MRF Meningococcus Genome Library, Sharif Shaaban, and Bingqing Zhu for providing CC4821 genomes through the Neisseria MLST Database. We thank Don Weiss of New York City Department of Health and Mental Hygiene for sharing with us the epidemiologic information of isolate S00-1268. We thank Xin Wang, Henju Marjuki, and Nadav Topaz of US Centers for Disease Control and Prevention for sharing with us the epidemiological information of isolates M29621, M38893, and M40156. We thank Zhaoyi Jia of Hebei CDC for checking the epidemiological information of isolate 130644. We thank Jay Lucidarme and Ray Borrow of Public Health England for sharing with us the epidemiological information of isolate M14 240580. We thank Hongyou Chen of Shanghai CDC for assistance of Gubbins analysis.
This study made use of Neisseria genomic data deposited in the Neisseria PubMLST Database (https://pubmlst.org/neisseria/) sited at the University of Oxford (Jolley & Maiden 2018, Wellcome open research, 3:124); the development of this database has been funded by the Wellcome Trust and European Union. This work was supported by National Natural Science Foundation of China under grant 81872909 and 81601801, Shanghai Rising-Star Program from Shanghai Municipal Science and Technology Commission (grant 17QA1403100), a Municipal Human Resources Development Program for Outstanding Young Talents in Medical and Health Sciences in Shanghai (grant 2017YQ039), and the 13th Five-Year Project of National Health and Family Planning Commission of the People’s Republic of China (grants 2017ZX10303405004 and 2017ZX10103009-003). The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
References
- Jafri RZ, Ali A, Messonnier NE, Tevi-Benissan C, Durrheim D, Eskola J, et al. Global epidemiology of invasive meningococcal disease. Popul Health Metr. 2013;11:17. DOIPubMedGoogle Scholar
- Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. DOIPubMedGoogle Scholar
- Zhou H, Gao Y, Xu L, Li M, Li Q, Li Y, et al. Distribution of serogroups and sequence types in disease-associated and carrier strains of Neisseria meningitidis isolated in China between 2003 and 2008. Epidemiol Infect. 2012;140:1296–303. DOIPubMedGoogle Scholar
- Shao Z, Li W, Ren J, Liang X, Xu L, Diao B, et al. Identification of a new Neisseria meningitidis serogroup C clone from Anhui province, China. Lancet. 2006;367:419–23. DOIPubMedGoogle Scholar
- Chen M, Guo Q, Wang Y, Zou Y, Wang G, Zhang X, et al. Shifts in the antibiotic susceptibility, serogroups, and clonal complexes of Neisseria meningitidis in Shanghai, China: a time trend analysis of the pre-quinolone and quinolone eras. PLoS Med. 2015;12:e1001838, discussion e1001838. DOIPubMedGoogle Scholar
- Guo Q, Mustapha MM, Chen M, Qu D, Zhang X, Chen M, et al. Evolution of sequence type 4821 clonal complex meningococcal strains in China from prequinolone to quinolone era, 1972–2013. Emerg Infect Dis. 2018;24:683–90. DOIPubMedGoogle Scholar
- Zhu B, Xu Z, Du P, Xu L, Sun X, Gao Y, et al. Sequence type 4821 clonal complex serogroup B Neisseria meningitidis in China, 1978–2013. Emerg Infect Dis. 2015;21:925–32. DOIPubMedGoogle Scholar
- Peng J, Yang L, Yang F, Yang J, Yan Y, Nie H, et al. Characterization of ST-4821 complex, a unique Neisseria meningitidis clone. Genomics. 2008;91:78–87. DOIPubMedGoogle Scholar
- Chiou CS, Liao JC, Liao TL, Li CC, Chou CY, Chang HL, et al. Molecular epidemiology and emergence of worldwide epidemic clones of Neisseria meningitidis in Taiwan. BMC Infect Dis. 2006;6:25. DOIPubMedGoogle Scholar
- Kawasaki Y, Matsubara K, Takahashi H, Morita M, Ohnishi M, Hori M, et al. Invasive meningococcal disease due to ciprofloxacin-resistant Neisseria meningitidis sequence type 4821: The first case in Japan. J Infect Chemother. 2018;24:305–8. DOIPubMedGoogle Scholar
- Tsang RS, Law DK, Deng S, Hoang L. Ciprofloxacin-resistant Neisseria meningitidis in Canada: likely imported strains. Can J Microbiol. 2017;63:265–8. DOIPubMedGoogle Scholar
- Harrison OB, Cole K, Peters J, Cresswell F, Dean G, Eyre DW, et al. Genomic analysis of urogenital and rectal Neisseria meningitidis isolates reveals encapsulated hyperinvasive meningococci and coincident multidrug-resistant gonococci. Sex Transm Infect. 2017;93:445–51. DOIPubMedGoogle Scholar
- Chen M, Wang W, Tu L, Zheng Y, Pan H, Wang G, et al. An emm5 Group A streptococcal outbreak among workers in a factory manufacturing telephone accessories. Front Microbiol. 2017;8:1156. DOIPubMedGoogle Scholar
- Topaz N, Boxrud D, Retchless AC, Nichols M, Chang HY, Hu F, et al. BMScan: using whole genome similarity to rapidly and accurately identify bacterial meningitis causing species. BMC Infect Dis. 2018;18:405. DOIPubMedGoogle Scholar
- Veeraraghavan B, Neeravi AR, Devanga Ragupathi NK, Inbanathan FY, Pragasam AK, Verghese VP. Whole-genome shotgun sequencing of the first observation of Neisseria meningitidis sequence type 6928 in India. Genome Announc. 2016;4:e01232–16. DOIPubMedGoogle Scholar
- Moura ARSS, Kretz CB, Ferreira IE, Nunes AMPB, de Moraes JC, Reis MG, et al. Molecular characterization of Neisseria meningitidis isolates recovered from 11-19-year-old meningococcal carriers in Salvador, Brazil. PLoS One. 2017;12:
e0185038 . DOIPubMedGoogle Scholar - Ezeoke I, Galac MR, Lin Y, Liem AT, Roth PA, Kilianski A, et al. Tracking a serial killer: Integrating phylogenetic relationships, epidemiology, and geography for two invasive meningococcal disease outbreaks. PLoS One. 2018;13:
e0202615 . DOIPubMedGoogle Scholar - Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. DOIPubMedGoogle Scholar
- Wu X, Li K, Xie M, Yu M, Tang S, Li Z, et al. Construction and protective immunogenicity of DNA vaccine pNMB0315 against Neisseria meningitidis serogroup B. Mol Med Rep. 2018;17:3178–85.PubMedGoogle Scholar
- Lewis LA, Ngampasutadol J, Wallace R, Reid JE, Vogel U, Ram S. The meningococcal vaccine candidate neisserial surface protein A (NspA) binds to factor H and enhances meningococcal resistance to complement. PLoS Pathog. 2010;6:
e1001027 . DOIPubMedGoogle Scholar - Poolman JT, Richmond P. Multivalent meningococcal serogroup B vaccines: challenges in predicting protection and measuring effectiveness. Expert Rev Vaccines. 2015;14:1277–87. DOIPubMedGoogle Scholar
- Fegan JE, Calmettes C, Islam EA, Ahn SK, Chaudhuri S, Yu RH, et al. Utility of hybrid transferrin binding protein antigens for protection against pathogenic Neisseria species. Front Immunol. 2019;10:247. DOIPubMedGoogle Scholar
- Unemo M, Shafer WM. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin Microbiol Rev. 2014;27:587–613. DOIPubMedGoogle Scholar
- Acevedo R, Bai X, Borrow R, Caugant DA, Carlos J, Ceyhan M, et al. The Global Meningococcal Initiative meeting on prevention of meningococcal disease worldwide: Epidemiology, surveillance, hypervirulent strains, antibiotic resistance and high-risk populations. Expert Rev Vaccines. 2019;18:15–30. DOIPubMedGoogle Scholar
- Rodrigues CMC, Jolley KA, Smith A, Cameron JC, Feavers IM, Maiden MCJ. Meningococcal deduced vaccine antigen reactivity (MenDeVAR) index: a rapid and accessible tool that exploits genomic data in public health and clinical microbiology applications. J Clin Microbiol. 2020;59:e02161–20. DOIPubMedGoogle Scholar
- Bratcher HB, Corton C, Jolley KA, Parkhill J, Maiden MCJ. A gene-by-gene population genomics platform: de novo assembly, annotation and genealogical analysis of 108 representative Neisseria meningitidis genomes. BMC Genomics. 2014;15:1138. DOIPubMedGoogle Scholar
- Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018;3:124. DOIPubMedGoogle Scholar
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. DOIPubMedGoogle Scholar
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(D1):D109–14. DOIPubMedGoogle Scholar
- Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524. DOIPubMedGoogle Scholar
- Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:
e15 . DOIPubMedGoogle Scholar - Mustapha MM, Marsh JW, Krauland MG, Fernandez JO, de Lemos AP, Dunning Hotopp JC, et al. Genomic investigation reveals highly conserved, mosaic, recombination events associated with capsular switching among invasive Neisseria meningitidis serogroup W sequence type (ST)-11 strains. Genome Biol Evol. 2016;8:2065–75. DOIPubMedGoogle Scholar
- Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. DOIPubMedGoogle Scholar
- Ladhani SN, Lucidarme J, Parikh SR, Campbell H, Borrow R, Ramsay ME. Meningococcal disease and sexual transmission: urogenital and anorectal infections and invasive disease due to Neisseria meningitidis. Lancet. 2020;395:1865–77. DOIPubMedGoogle Scholar
- Harrison LH, Trotter CL, Ramsay ME. Global epidemiology of meningococcal disease. Vaccine. 2009;27(Suppl 2):B51–63. DOIPubMedGoogle Scholar
- Krone M, Gray S, Abad R, Skoczyńska A, Stefanelli P, van der Ende A, et al. Increase of invasive meningococcal serogroup W disease in Europe, 2013 to 2017. Euro Surveill. 2019;24:24. DOIPubMedGoogle Scholar
- Lucidarme J, Hill DM, Bratcher HB, Gray SJ, du Plessis M, Tsang RS, et al. Genomic resolution of an aggressive, widespread, diverse and expanding meningococcal serogroup B, C and W lineage. J Infect. 2015;71:544–52. DOIPubMedGoogle Scholar
- Mayer LW, Reeves MW, Al-Hamdan N, Sacchi CT, Taha MK, Ajello GW, et al. Outbreak of W135 meningococcal disease in 2000: not emergence of a new W135 strain but clonal expansion within the electophoretic type-37 complex. J Infect Dis. 2002;185:1596–605. DOIPubMedGoogle Scholar
- Veeraraghavan B, Lal B, Devanga Ragupathi NK, Neeravi IR, Jeyaraman R, Varghese R, et al. First genome report on novel sequence types of Neisseria meningitidis: ST12777 and ST12778. J Glob Antimicrob Resist. 2018;12:117–8. DOIPubMedGoogle Scholar
- Schoen C, Kischkies L, Elias J, Ampattu BJ. Metabolism and virulence in Neisseria meningitidis. Front Cell Infect Microbiol. 2014;4:114. DOIPubMedGoogle Scholar
- Watkins ER, Maiden MC. Metabolic shift in the emergence of hyperinvasive pandemic meningococcal lineages. Sci Rep. 2017;7:41126. DOIPubMedGoogle Scholar
- Chen M, Rodrigues CMC, Harrison OB, Zhang C, Tan T, Chen J, et al. Invasive meningococcal disease in Shanghai, China from 1950 to 2016: implications for serogroup B vaccine implementation. Sci Rep. 2018;8:12334. DOIPubMedGoogle Scholar
- Adriaenssens N, Coenen S, Kroes AC, Versporten A, Vankerckhoven V, Muller A, et al.; ESAC Project Group. European Surveillance of Antimicrobial Consumption (ESAC): systemic antiviral use in Europe. J Antimicrob Chemother. 2011;66(Suppl 6):1897–905. DOIPubMedGoogle Scholar
- Van Boeckel TP, Gandra S, Ashok A, Caudron Q, Grenfell BT, Levin SA, et al. Global antibiotic consumption 2000 to 2010: an analysis of national pharmaceutical sales data. Lancet Infect Dis. 2014;14:742–50. DOIPubMedGoogle Scholar
- Taha MK, Vázquez JA, Hong E, Bennett DE, Bertrand S, Bukovski S, et al. Target gene sequencing to characterize the penicillin G susceptibility of Neisseria meningitidis. Antimicrob Agents Chemother. 2007;51:2784–92. DOIPubMedGoogle Scholar
- Chen M, Zhang C, Zhang X, Chen M. meningococcal quinolone resistance originated from several commensal Neisseria species. Antimicrob Agents Chemother. 2020;64:e01494–19.PubMedGoogle Scholar
- Deghmane AE, Hong E, Taha MK. Emergence of meningococci with reduced susceptibility to third-generation cephalosporins. J Antimicrob Chemother. 2017;72:95–8. DOIPubMedGoogle Scholar
- Li J, Shao Z, Liu G, Bai X, Borrow R, Chen M, et al. Meningococcal disease and control in China: Findings and updates from the Global Meningococcal Initiative (GMI). J Infect. 2018;76:429–37. DOIPubMedGoogle Scholar
- Ladhani SN, Ramsay M, Borrow R, Riordan A, Watson JM, Pollard AJ. Enter B and W: two new meningococcal vaccine programmes launched. Arch Dis Child. 2016;101:91–5. DOIPubMedGoogle Scholar
- Ostergaard L, Vesikari T, Absalon J, Beeslaar J, Ward BJ, Senders S, et al.; B1971009 and B1971016 Trial Investigators. B1971009 and B1971016 Trial Investigators. A bivalent meningococcal B vaccine in adolescents and young adults. N Engl J Med. 2017;377:2349–62. DOIPubMedGoogle Scholar
Figures
Tables
Cite This ArticleOriginal Publication Date: March 04, 2021
1These authors contributed equally to this article.
Table of Contents – Volume 27, Number 4—April 2021
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Mingliang Chen or Min Chen, Shanghai Municipal Center for Disease Control and Prevention, 1380 West ZhongShan Rd, Shanghai 200336, China
Top