Volume 29, Number 1—January 2023
Dispatch
Genomic Microevolution of Vibrio cholerae O1, Lake Tanganyika Basin, Africa
Abstract
Africa’s Lake Tanganyika basin is a cholera hotspot. During 2001–2020, Vibrio cholerae O1 isolates obtained from the Democratic Republic of the Congo side of the lake belonged to 2 of the 5 clades of the AFR10 sublineage. One clade became predominant after acquiring a parC mutation that decreased susceptibility to ciprofloxacin.
Cholera is an acute life-threatening diarrheal disease responsible for ≈4.3 million cases and 142,000 deaths annually worldwide (1). Excluding epidemic peaks in Haiti and Yemen (2,3), most cases of cholera originate from sub-Saharan Africa, predominantly the African Great Lakes Region (AGLR); specifically, the countries of the Lake Tanganyika basin (4). Many recurrent cholera outbreaks in the Democratic Republic of the Congo (DRC), Tanzania, Burundi, and Zambia have been linked to a common hotspot area around the Lake Tanganyika basin (5–8).
By the end of 2018, the World Health Organization had noted a steady decline in cholera cases throughout the world, including the AGLR (9). Continuous genomic surveillance of circulating Vibrio cholerae bacteria strains is required to understand the transmission dynamics and genetic evolution of V. cholerae and potentially to guide prevention and response interventions to continue the trend toward decreasing case numbers, in line with the global cholera roadmap to 2030 (10). One lineage, seventh pandemic V. cholerae O1 El Tor (7PET), is responsible for the current pandemic, which began in 1961 (11); Africa was hit by 7PET in 1970 (11). During 1970–2014, >11 different 7PET sublineages were introduced from South Asia into Africa, and sublineage AFR10 (previously T10) replaced AFR5 (previously T5) in the AGLR in the late 1990s (11). Sublineage AFR13 (previously T13) was identified in East Africa (Tanzania, Uganda, Kenya) and Zimbabwe (12). We tracked the 7PET populations circulating in the Lake Tanganyika basin by studying recent V. cholerae O1 isolates collected in the region by conventional bacteriology and genomics and placing these genomes in a broader phylogenetic context to elucidate their evolutionary history.
Figure 1
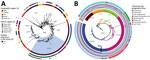
Figure 1. Phylogenomics of clinical and environmental Vibrio cholerae O1 El Tor isolates from the Lake Tanganyika basin, Africa. A) Maximum-likelihood phylogeny of 1,366 seventh pandemic V. choleraeO1...
We analyzed 96 V. cholerae O1 isolates collected during 2015–2020 in DRC (86 clinical isolates, including 39 collected in 2018–2020) and Tanzania (10 environmental isolates from fish and lake water) (Appendix 1; Appendix 2 Table 1). We subjected the isolates to antimicrobial susceptibility testing, whole-genome sequencing, genomic characterization, and phylogenetic analyses, as previously described (11,12) (Appendix 1). We performed a phylogenetic analysis of these genomes within a global collection of 1,366 7PET V. cholerae O1 genomes (Appendix 2 Table 2), including another 130 genomes from DRC collected during 1984–2017. We based the final maximum-likelihood phylogenetic tree on 10,352 single-nucleotide variants distributed over the nonrepetitive, nonrecombinant core genome (Figure 1, panel A).
Figure 2
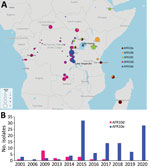
Figure 2. A) Spatiotemporal dynamics of the AFR10 clades of Vibrio choleraeO1 in the African Great Lakes Region, Africa, 1998–2020. Circle size indicates the number of isolates at the location...
Phylogenetic analysis of the 96 genomes of V. cholerae O1 isolates showed that all belonged to 7PET sublineage AFR10 (Figure 1, panel A). Within the limits of our sampling, sublineage AFR5, which circulated actively in the AGLR during the 1980s–1990s (11), appears to be extinct in the region, whereas sublineage AFR13, reported in 2015 in Uganda and Tanzania (12), has not yet spread to DRC. Since 1998, the endemicity of the AFR10 sublineage in the Tanganyika basin and surrounding countries has led to microevolution; an analysis of 357 AFR10 genomes from our global dataset revealed the presence of 5 clades, AFR10a–AFR10e (Figure 1, panel B; Appendix 1 Figure 1). Clades AFR10a, AFR10b, and AFR10c were mostly associated with the eastern AGLR countries. The isolates of these clades were of sequence type (ST) 69 (Figure 1, panel B). Clades AFR10d and AFR10e predominated in DRC and the Lake Tanganyika basin. Clade AFR10d is of ST515, essentially Inaba serotype, and was widespread in DRC and neighboring countries (Figure 2, panel A), as previously reported (13). It was the only clade found in the Lake Kivu and Lake Edward basins. AFR10e strains are of ST69, Ogawa serotype, and were essentially restricted to the Lake Tanganyika basin, confirming previous findings (13). A further pangenome analysis of the AFR10 isolates revealed no clade-specific gain or loss of genes (Appendix 1 Figure 2). AFR10e strains have gradually replaced AFR10d strains in the region since 2014; all V. cholerae O1 strains obtained from the Tanganyika basin by 2017, as well as those obtained from the lake itself in 2018 and 2019, were AFR10e strains (Figure 2 panel B). Epidemiologic studies identified cholera hotspots in the AGLR as a source of major countrywide outbreaks reaching the capital, Kinshasa, and the Atlantic coast, via the Congo River, in 2011, 2012, and 2016 (5). These outbreaks were caused primarily by clade AFR10d, which has a wider geographic distribution than clade AFR10e (Figure 2) (14).
One striking characteristic of AFR10e isolates was the presence of a mutation in the quinolone resistance–determining region of the topoisomerase IV subunit A gene, parC (S85L), that led to higher MIC values (0.25–0.38 mg/L) for ciprofloxacin. These isolates, which have a lower susceptibility to ciprofloxacin than wild-type populations, would be classified as either resistant (MIC 0.38 mg/L) or susceptible (MIC 0.25 mg/L) in accordance with clinical breakpoints published by the European Committee on Antimicrobial Susceptibility Testing (https://www.eucast.org/clinical_breakpoints) in 2022, as inferred from the study of Vibrio strains and not Enterobacteriaceae strains. This parC mutation, sporadically reported in other AFR10 clades (Table; Figure 1, panel B; Appendix 2 Table 1), has been a distinctive characteristic of AFR10e isolates since 2014 (Figure 2, panel B). It was the second mutation affecting susceptibility to quinolones and fluroquinolones to be found in this AFR10e clade; the first was a mutation in the DNA gyrase subunit A gene, gyrA (S83I), present in all AFR10 isolates. This additional mutation does not seem to be associated with the specific use of fluoroquinolones for treating cholera outbreaks, because the antimicrobial drugs used for first-line cholera control in DRC are tetracyclines and macrolides, to which AFR10e isolates remain susceptible. Instead, the mutation may result from widespread self-medication with antimicrobials, a common practice in many sub-Saharan Africa countries including DRC (15).
All 96 isolates analyzed had known mutations of the VC_0715 and VC_A0637 genes conferring nitrofuran resistance (Table), consistent with previous findings (2). The isolates also carried the SXT/R391 genomic element ICEVchInd5, encoding resistance to streptomycin (strAB), sulfonamides (sul2), chloramphenicol (floR), trimethoprim and the O/129 vibriostatic agent (dfrA1), and trimethoprim–sulfamethoxazole (sul2 and dfrA1), with concordance between the phenotypic and genotypic data (Table; Appendix 2 Table 1).
We found that the cholera outbreaks in the eastern part of DRC during 2001–2020 were caused by V. cholerae O1 sublineage AFR10, which was introduced into East Africa from South Asia in the late 1990s. The AFR13 sublineage was already reported in 2015 in Tanzania, including the city of Kigoma, was located on the shore of Lake Tanganyika but had not been detected in DRC as of 2022. The AFR10 isolates of this region belong principally to 2 clades, AFR10d (Inaba, ST515) and AFR10e (Ogawa, ST69). AFR10d was responsible for outbreaks reported in the western part of DRC in 2011–2017 and neighboring countries; AFR10e (Ogawa, ST69) was restricted to the Lake Tanganyika basin, in which reduced susceptibility to ciprofloxacin has been seen since 2014. Lake Tanganyika seems to serve as a transmission channel, favoring the establishment of AFR10e in local human populations. Further investigation, including studies of population movement, should reveal why AFR10e clade has remained within the Lake Tanganyika basin. The replacement of other clades by this antimicrobial-resistant clade in this area highlights the need for more systematic documentation of antimicrobial drug use and the implementation of adapted stewardship programs, particularly in outbreak responses. Overall, these findings highlight the need for continuous genomic surveillance and for coordinated communication between countries for effective interventions.
Dr. Hounmanou is a postdoctoral fellow specializing in One Health at the University of Copenhagen. His primary research interests are microbial genomics, antimicrobial resistance, and routes of transmission between animals, humans, and bodies of water.
Acknowledgment
The International Foundation for Sciences funded the fieldwork for the isolation of environmental isolates (grant no. 1-2-A-6100-1). A.D.’s laboratory work is supported by core funding from the University of Copenhagen. The London School of Hygiene and Tropical Medicine study was cofunded by the French Agency for Development and the Veolia Foundation. The laboratory of F.-X.W. is part of the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence funded by the Government of France’s “Investissement d'Avenir” programme (grant no. ANR-10-LABX-62-IBEID). The CNRVC is cofunded by Santé Publique France and the Institut Pasteur.
References
- Weil AA, Ryan ET. Cholera: recent updates. Curr Opin Infect Dis. 2018;31:455–61. DOIPubMedGoogle Scholar
- Weill FX, Domman D, Njamkepo E, Almesbahi AA, Naji M, Nasher SS, et al. Genomic insights into the 2016-2017 cholera epidemic in Yemen. Nature. 2019;565:230–3. DOIPubMedGoogle Scholar
- Hendriksen RS, Price LB, Schupp JM, Gillece JD, Kaas RS, Engelthaler DM, et al. Population genetics of Vibrio cholerae from Nepal in 2010: evidence on the origin of the Haitian outbreak. MBio. 2011;2:e00157–11. DOIPubMedGoogle Scholar
- Lessler J, Moore SM, Luquero FJ, McKay HS, Grais R, Henkens M, et al. Mapping the burden of cholera in sub-Saharan Africa and implications for control: an analysis of data across geographical scales. Lancet. 2018;391:1908–15. DOIPubMedGoogle Scholar
- Ingelbeen B, Hendrickx D, Miwanda B, van der Sande MAB, Mossoko M, Vochten H, et al. Recurrent cholera outbreaks, Democratic Republic of the Congo, 2008–2017. Emerg Infect Dis. 2019;25:856–64. DOIPubMedGoogle Scholar
- Hounmanou YMG, Mølbak K, Kähler J, Mdegela RH, Olsen JE, Dalsgaard A. Cholera hotspots and surveillance constraints contributing to recurrent epidemics in Tanzania. BMC Res Notes. 2019;12:664. DOIPubMedGoogle Scholar
- Debes AK, Shaffer AM, Ndikumana T, Liesse I, Ribaira E, Djumo C, et al. Cholera hot-spots and contextual factors in Burundi, planning for elimination. Trop Med Infect Dis. 2021;6:76. DOIPubMedGoogle Scholar
- Mwaba J, Debes AK, Shea P, Mukonka V, Chewe O, Chisenga C, et al. Identification of cholera hotspots in Zambia: A spatiotemporal analysis of cholera data from 2008 to 2017. PLoS Negl Trop Dis. 2020;14:
e0008227 . DOIPubMedGoogle Scholar - World Health Organization. Drop in cholera cases worldwide, as key endemic countries report gains in cholera control. 2019 [cited 2020 Apr 22]. https://www.who.int/news-room/detail/19-12-2019-drop-in-cholera-cases-worldwide-as-key-endemic-countries-report-gains-in-cholera-control
- World Health Organization. Ending cholera. a global roadmap to 2030. 2017 [cited 2020 Apr 22]. https://www.gtfcc.org/wp-content/uploads/2019/10/gtfcc-ending-cholera-a-global-roadmap-to-2030.pdf
- Weill FX, Domman D, Njamkepo E, Tarr C, Rauzier J, Fawal N, et al. Genomic history of the seventh pandemic of cholera in Africa. Science. 2017;358:785–9. DOIPubMedGoogle Scholar
- Hounmanou YMG, Leekitcharoenphon P, Kudirkiene E, Mdegela RH, Hendriksen RS, Olsen JE, et al. Genomic insights into Vibrio cholerae O1 responsible for cholera epidemics in Tanzania between 1993 and 2017. PLoS Negl Trop Dis. 2019;13:
e0007934 . DOIPubMedGoogle Scholar - Irenge LM, Ambroise J, Mitangala PN, Bearzatto B, Kabangwa RKS, Durant JF, et al. Genomic analysis of pathogenic isolates of Vibrio cholerae from eastern Democratic Republic of the Congo (2014-2017). PLoS Negl Trop Dis. 2020;14:
e0007642 . DOIPubMedGoogle Scholar - Breurec S, Franck T, Njamkepo E, Mbecko JR, Rauzier J, Sanke-Waïgana H, et al. Seventh Pandemic Vibrio cholerae O1 sublineages, Central African Republic. Emerg Infect Dis. 2021;27:262–6. DOIPubMedGoogle Scholar
- Belachew SA, Hall L, Selvey LA. Non-prescription dispensing of antibiotic agents among community drug retail outlets in Sub-Saharan African countries: a systematic review and meta-analysis. Antimicrob Resist Infect Control. 2021;10:13. DOIPubMedGoogle Scholar
Figures
Table
Cite This ArticleTable of Contents – Volume 29, Number 1—January 2023
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Marie-Laure Quilici, Institut Pasteur, Unité des Bactéries Pathogènes Entériques, 28 rue du Dr. Roux, 75724 Paris CEDEX 15, France
Top