Volume 22, Number 8—August 2016
Research
Phylogeographic Evidence for 2 Genetically Distinct Zoonotic Plasmodium knowlesi Parasites, Malaysia
Abstract
Infections of humans with the zoonotic simian malaria parasite Plasmodium knowlesi occur throughout Southeast Asia, although most cases have occurred in Malaysia, where P. knowlesi is now the dominant malaria species. This apparently skewed distribution prompted an investigation of the phylogeography of this parasite in 2 geographically separated regions of Malaysia, Peninsular Malaysia and Malaysian Borneo. We investigated samples collected from humans and macaques in these regions. Haplotype network analyses of sequences from 2 P. knowlesi genes, type A small subunit ribosomal 18S RNA and cytochrome c oxidase subunit I, showed 2 genetically distinct divergent clusters, 1 from each of the 2 regions of Malaysia. We propose that these parasites represent 2 distinct P. knowlesi types that independently became zoonotic. These types would have evolved after the sea-level rise at the end of the last ice age, which separated Malaysian Borneo from Peninsular Malaysia.
The number of malaria cases in Malaysia steadily decreased from a peak of 59,208 in 1995 to 3,850 confirmed cases in 2013; of these, 80% were reported in the 2 states of Malaysian Borneo and the remainder in 6 of the 11 states of Peninsular Malaysia (Figure 1) (1). In Malaysia, the simian malarial parasite species Plasmodium knowlesi is now the dominant species infecting humans and is >2 times more prevalent than P. falciparum or P. vivax. Humans were found to be susceptible to P. knowlesi when this species was experimentally transmitted to man in 1932, the year in which it was first described (2,3). In 1965, the first confirmed case of a naturally acquired infection in humans was recorded (4). The next naturally acquired confirmed cases were reported in 2004, when a stable focus of P. knowlesi was discovered in Sarawak, 1 of 2 states that make up Malaysian Borneo (5). Thereafter, transmission of P. knowlesi to humans occurred in the second state, Sabah (6,7), and in neighboring countries (8,9).
The natural hosts of P. knowlesi are principally the long-tailed (Macaca fascicularis) and pig-tailed (M. nemestrina) macaques (10), 2 species that are widely distributed in the Southeast Asia countries in which cases of P. knowlesi have been recorded. To date, human-to-human transmission has not been observed. Infections in humans can cause severe disease that can be fatal (9,11), underscoring the public health concern raised by this zoonotic simian parasite.
The 2 states of Malaysian Borneo appear to be the epicenter of zoonotic P. knowlesi infections: 1,391 cases in Malaysian Borneo and 423 cases in Peninsular Malaysia were recorded in 2012. A total of 1,407 PCR-confirmed cases were reported during 2004–2013 in Malaysia, which contrasts with the low number of cases (n = 136) reported from neighboring countries (9): Cambodia (n = 1), China (n = 36), Indonesia (n = 1), Myanmar (n = 14), the Philippines (n = 5), Singapore (n = 2), Thailand (n = 36), and Vietnam (n = 32). The reasons for this uneven distribution remain unclear. Geographic variation in mosquito species and human social factors could be an explanation; it is also possible that the parasite populations circulating on the island of Borneo are distinct from those found in continental Malaysia. The P. knowlesi strains that had been studied in earlier years displayed distinct biologic characteristics, which in some cases led malariologists to propose distinct P. knowlesi subspecies (12). Such differences could indicate that local ecologic factors are influential and that each P. knowlesi subspecies became a zoonosis independently in each geographic area.
To explore whether the P. knowlesi populations in Malaysia differed and independently became zoonoses, we focused on 2 genes that have been extensively used for phylogenetic studies (13–16): 1 nuclear, encoding the type A small subunit ribosomal 18S RNA (PkA-type 18S rRNA), and 1 mitochondrial, encoding the cytochrome oxidase subunit I protein (PkCOX1). Using samples collected from humans and macaques in both regions of the country, we generated the relevant sequences, compared them to those published previously (17), and conducted phylogenetic and population genetic analyses.
Sample Collection
The Medical Research Ethic Committee of the Ministry of Health Malaysia, Sabah State Director of Health, Kelantan State Director of Health, and directors of state and district hospitals approved this study. Ethical approval was granted by the Medical Research and Ethics Committee of the Malaysian Ministry of Health (Reference Number: KKM/NIHSEC/800/-2/2/2/P13–316), the Medical Ethics Committee of University Malaya Medical Centre, and the Department of Wildlife and National Parks.
Figure 1
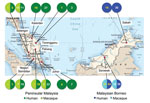
Figure 1. Geographic origin of the genetic sequences generated during study of Plasmodium knowlesi parasite populations, Malaysia. The numbers in each circle refer to the number of sequences (macaque or human) obtained for...
We used previously collected human blood samples for this study (7): 78 microscopically confirmed P. knowlesi–positive blood samples from patients in 8 states in Malaysia, including Sabah and Sarawak in Malaysian Borneo (Figure 1). We also examined blood samples from 8 long-tailed macaques collected during routine surveys by the Department of Wildlife and National Parks in the Peninsular Malaysia states of Pahang, Selangor, and Negeri Sembilan. All samples were collected during September 2012–December 2013 (Tables 1, 2). In addition to these samples, we included previously published sequences deposited into GenBank during 2003–2015 in the analyses; these sequences were derived from samples collected from humans and macaques (Technical Appendix Table).
Amplification and Sequencing of Gene Fragments
Genomic DNA was extracted from the human and macaque blood samples by using the DNeasy Blood Tissue Kit (QIAGEN, Hilden, Germany), according to the manufacturers’ protocol. An established nested PCR protocol was used to test the samples; all tested positive for P. knowlesi only (5).
The PkA-type 18S rRNA and PkCOX1 genes were then amplified (MyCycler, Bio-Rad, Hercules, CA). In a primary amplification reaction, a PkA-type 18S rRNA fragment of 1.1. kb was obtained by using the oligonucleotide primer pair rPLU5+rPLU6 (18). The amplification reaction was completed in a mixture containing 1X Green Go Taq Flexi Buffer (Promega, Madison, WI, USA); 4.0 mol/Lmagnesium chloride solution; 0.2 mol/L dNTP Mix (Promega), 0.2 μM of each primer; 1 U GoTaq Flexi DNA Polymerase (Promega); and 4 μL of DNA template combined with nuclease–free water to obtain a final volume of 25 μL. The PCR amplification was initiated at 95°C for 10 min, then by 35 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, extension at 72°C for 1 min, and a final extension at 72°C for 5 min. For the secondary amplification, 4 μL of the primary amplification product were used as a template by using forward primer Pkl1 (5′-ACATAACTGATGCCTCCGCGTA) and reverse primer Pkl2 (5′-CACACATCGTTCCTCTAAGAAGC) to obtain a 986–990-bp fragment. The reaction mixture and the cycling conditions were as above with minor modification for the 35 cycles: denaturation at 94°C for 1 min, annealing at 53°C for 1 min, extension at 72°C for 1 min, and final extension at 72°C for 10 min. The amplified PCR fragments were cloned into the pGEM-T Vector (Invitrogen, Carlsbad, CA, USA). Plasmids purified from >2 positive clones from each ligation mixture were selected for sequencing (First Base Laboratories Sdn Bhd, Malaysia). Any polymorphism that was not observed in >2 samples was only included in the analysis if its validity was confirmed by a repeated cycle of amplification, cloning, and sequencing.
For the PkCOX1 gene, amplification was achieved by using forward (5′-GCCAGGATTATTTGGAGG) and reverse (5′-CAGGAATACGTCTAGGCA) primers to obtain a 1,116-bp fragment. These primers were designed based on a published gene sequence (GenBank accession no. AY598141). The amplification reaction was achieved as above. The PCR amplification was initiated at 95°C for 3 min, then denatured for 35 cycles at 94°C for 1 min, annealed at 52°C for 1 min, extended at 72°C for 1 minute, and put through final extension at 72°C for 10 min. The purified amplified fragments were then sent for sequencing.
Sequence Editing and Alignment
We analyzed the DNA sequences using BioEdit Sequence Alignment Editor Software (http://www.mbio.ncsu.edu/BioEdit/bioedit.html) on the reference P. knowlesi H-strain (GenBank accession no. AM910985) for the PkA-type 18S rRNA and the P. knowlesi mitochondrial sequence (GenBank accession no. NC 00723244) for the PkCOX1 gene. Results were exported to MEGA 5.6 software (http://www.megasoftware.net) for further alignment and analysis. We performed similarity searches using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). We obtained 28 additional PkA-type 18S RNA sequences derived from P. knowlesi–infected samples (9 from humans and 19 from macaques) and 46 additional PkCOX1 sequences derived from P. knowlesi-infected samples (22 from humans and 24 from macaques) from GenBank and included these sequences in the analysis (online Technical Appendix Table).
Haplotype Network Analysis
We estimated polymorphism of the PkA-type 18S rRNA and PkCOX1 genes by computing haplotype diversity (Hd), number of haplotypes (h), nucleotide diversity (π), number of polymorphic sites, and the average number of pairwise nucleotide differences using DnaSP version 5.10.01 software (BioSoft http://en.bio-soft.net/). We constructed haplotype networks for PkA-type 18S rRNA and PkCOX1 genes based on their polymorphic sites by using the median-joining method in NETWORK version 4.6.1.2 software (Fluxus Technology Ltd, Suffolk, UK). We inferred the genealogical haplotype network using the sequences of P. knowlesi human and macaque isolates from Peninsular Malaysia and Malaysian Borneo. Where available, we included sequences from the P. knowlesi H and Nuri strains as references.
Population Genetic Structure Analysis
To define genetic structure of the P. knowlesi parasite population in Malaysia, we used STRUCTURE version 2.3.4 software (The Pritchard Lab, Stanford University, Stanford, CA, USA) that deploys the Bayesian model–based clustering approach. We estimated the most probable number of populations (K) using an admixture model. All sample data (for both genes) were run for values K = 1–8, each with a total of 15 iterations. We used 500,000 Markov Chain Monte Carlo generations for each run after a burn-in of 50,000 steps. The most likely number K in the data was estimated by calculating ΔK values and identifying the K value that maximizes the log probability of data, lnP(D) (19). The most probable K value was then calculated according to Evanno’s method (20) by using the webpage interface STRUCTURE Harvester (21). We also used ARLEQUIN version 3.5.1.3 software (University of Berne, Berne, Switzerland) to compute pairwise differences (FST) between populations (i.e., humans and macaques from Peninsular Malaysia and Malaysian Borneo) (22) from haplotypes that showed 10,100 permutations. FST is a comparison of the sum of genetic variability within and between populations on the basis of the differences in allelic frequencies. We interpreted FST values as no (0), low (>0–0.05), moderate (0.05–0.15), and high (0.15–0.25) genetic differentiation.
Neutrality and Demographic Analysis
We examined departure from a strict neutral model, including demographic expansions, on the basis of pairwise mismatch distribution, the Tajima D test (23), Fu and Li D (24), Fu and Li F, and Fu Fs statistics (25) using DnaSP version. 5.10.01 software (26). Significant negative values for these tests indicate either a purifying selection or population expansion, and positive values indicate balancing selection.
The sequences analyzed in this study were derived from 130 P. knowlesi–infected blood samples obtained from 23 macaques and 107 humans. We analyzed a total of 209 PkA-type 18S RNA sequences (105 from Peninsular Malaysia and 104 from Malaysian Borneo) and 138 PkCOX1 sequences (54 from Peninsular Malaysia and 84 from Malaysian Borneo).
Gene Diversity Indices
Analysis of the molecular polymorphism within the 209 partial PkA-type 18S rRNA sequences (945 bp) revealed moderately polymorphic sequences (π = 0.00324 ± 0.00019). Overall, 137 polymorphic sites yielded 93 haplotypes. Nucleotide and haplotype diversities were broadly similar for both Peninsular Malaysia and Malaysian Borneo samples (Table 3). Single-nucleotide polymorphisms were scattered throughout the gene; most the Peninsular Malaysia sequences displayed a distinct single nucleotide polymorphism (G→A at position 830) (Technical Appendix Table).
Analysis of the molecular polymorphism within the 138 partial P. knowlesi mitochondrial COX1 sequences (1,082 bp) revealed low instances of polymorphism (π = 0.00215 + 0.00013). Overall, 61 polymorphic sites yielded 44 haplotypes. Although nucleotide diversities were similar for sequences from both regions, haplotype diversity was higher for the sequences from Peninsular Malaysia (h = 19, Hd = 0.827 ± 0.039) than for those from Malaysian Borneo (h = 25, Hd = 0.676 ± 0.057) (Table 3). Sequences from Malaysian Borneo were distinguished from those from Peninsular Malaysia by 2 distinct single nucleotide polymorphisms (G→A at position 166 andT→C at position 659) (Technical Appendix Figure).
Haplotype network
Figure 2
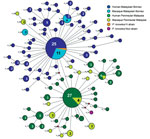
Figure 2. Median-joining networks of Plasmodium knowlesi type A small subunit ribosomal 18S RNA haplotypes from Malaysia. The genealogical haplotype network shows the relationships among the 93 haplotypes present in the 209 sequences...
Figure 3
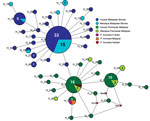
Figure 3. Median-joining networks of Plasmodium knowlesi cytochrome oxidase subunit I haplotypes from Malaysia. The genealogical haplotype network shows the relationships among the 44 haplotypes present in the 138 sequences obtained from human...
DNA sequence variation in our phylogeographic study is more clearly observed in a haplotype network. The network tree for the PkA-type 18S RNA haplotypes (Figure 2) showed 2 distinct P. knowlesi populations that, with 1 exception, clustered exclusively to 1 of the 2 regions of Malaysia: Peninsular Malaysia (n = 47) and Malaysian Borneo (n = 48). The exception was of 2 haplotypes derived from 1 macaque sample (haplotypes 88 and 89) from Peninsular Malaysia that clustered with the Malaysian Borneo haplotypes. In each cluster, only 2 haplotypes were shared by humans and macaques, but in each case 1 was dominant (in Malaysian Borneo, haplotype 1: n = 38, human = 27, macaque = 11; and in Peninsular Malaysia, haplotype 9: n = 31, human = 27, macaque = 4). The network tree for the PkCOX1 genes showed a similar pattern to that of PkA-type 18S rRNA: it had 2 geographically distinct P. knowlesi populations (n = 19 for Peninsular Malaysia and n = 25 for Malaysian Borneo) (Figure 3), and dominant haplotypes in each cluster were shared between humans and macaques (Figure 3).
Figure 4
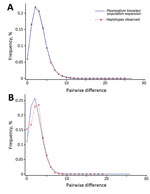
Figure 4. Pairwise mismatch distribution of Plasmodium knowlesi parasite populations, Malaysia. A) Type A small subunit ribosomal 18S RNA; B) cytochrome oxidase subunit I. Red dotted lines represent the observed frequencies of the...
The excess of unique PkA-type 18S rRNA and PkCOX1 haplotypes observed for the P. knowlesi populations in humans (Figures 2, 3) is indicative of an evolutionarily recent population expansion. A signature of population expansion was also evident from the unimodal shape of the pairwise mismatch distribution of the PkA-type 18S rRNA and PkCOX1 genes (Figure 4). Calculations by using Tajima D, Fu and Li D and F, and Fu Fs statistics also showed significant negative values (p = 0.05–0.001; Table 4). However, the low number of samples and consequent sequences from macaques precludes any meaningful comparison.
Population Structure
Figure 5

Figure 5. Most likely number of Plasmodium knowlesi parasite subpopulation haplotype clusters (K) in Malaysian Borneo (gray) and Peninsular Malaysia (white). A) Type A small subunit ribosomal 18S RNA (K = 2, ΔK...
We used a Bayesian admixture model implemented in STRUCTURE to calculate the potential number of P. knowlesi parasite populations within Malaysia. Because the study samples were collected from 8 different states of Malaysia (Figure 1), we used K values from 1 to 8 for the analysis. For both genes, significant genetic structure was found between the parasite populations when K = 2 (Figure 5), indicating 2 distinct populations clustered to 1 of the 2 main regions of Malaysia (PkA-type 18S rRNA, K = 2, ΔK = 121.79; PkCOX1, K = 2, ΔK = 481.27). In PkA-type 18S rRNA and PkCOX1 sequences, we also estimated pairwise FST values using ARLEQUIN software to determine to what extent population differentiation exists within P. knowlesi in Malaysia on the basis of host and geographic origin, i.e., between humans and macaques and between Peninsular Malaysia and Malaysian Borneo. This analysis revealed particularly high population differentiation FST values (>0.21 for PkA-type 18S rRNA and >0.60 for PkCOX1) for samples originating from Peninsular Malaysia and Malaysian Borneo irrespective of the host (human or macaque) from which they were collected (Table 5). For the macaque and human population within the same geographic region, the FST values were low (<0.05) (Table 5), suggesting that parasitic transmission was confined to each of the regions. These results are concordant with the haplotype network analysis.
The results of the various analyses conducted on the P. knowlesi parasites collected from Peninsular Malaysia and Malaysian Borneo strongly support the conclusion that the 2 geographically separated regions of this country harbor genetically distinct P. knowlesi populations. Haplotype diversity, a measure of species evenness (low values indicate skewing toward a few predominant haplotypes), was high for both the Peninsular Malaysia (0.906 ± 0. 0.025) and the Malaysian Borneo (0.871 ± 0.031) isolates, which may indicate a sustained transmission of P. knowlesi in both regions of Malaysia over long periods. Similar high haplotype diversity values have been reported for the P. knowlesi csp gene in isolates from Sarawak (17). Nucleotide diversity, however, was low for the genes analyzed in this study, irrespective of the samples’ geographic or host origins, indicating that only minor differences occurred between the haplotypes observed (Technical Appendix Table, Figure). A similar pattern has been observed for geographically separated P. vivax populations between which gene flow is limited (14).
The relatively large sample group size and the short genetic distances between the intraspecific sequences in this study are not suited for phylogenetic studies that aim to reconstruct genealogies. Therefore, we subjected the sequences to a median-joining haplotype network analysis, which showed that the network consisted mostly of unique haplotypes that clearly form 2 clusters: 1 comprised the samples obtained from Peninsular Malaysia, and the other the samples from Malaysian Borneo (Figures 2, 3). Within each cluster, the dominant haplotypes were shared between humans and macaques; a similar observation was previously reported for a sample set collected in Sarawak (17). Additional population structure analyses showed very high genetic differentiation between 2 distinct P. knowlesi populations from the 2 geographic regions and very low genetic differentiation between the human and macaque parasites within each of these regions (Figure 5). These observations strongly support the conclusion that humans are susceptible to infection by any of the P. knowlesi types circulating in macaques.
The question arises as to how these 2 distinct populations arose. Analyses of the complete mitochondrial DNA revealed that P. knowlesi parasites were present in macaques around 65,000 years ago, before human settlement in Southeast Asia (17). Although DNA sequences isolated in our study are too short to perform a comparable analysis, it has been proposed that the macaque populations of Borneo became isolated from those of Peninsular Malaysia, Java, and Sumatra around 15,000 years ago, when the rise in the level of the South China Sea at the end of the last ice age submerged parts of Sundaland (27). Thus, P. knowlesi populations likely became isolated, along with their natural vertebrate and insect hosts, and consequently evolved separately. This mechanism of geographic isolation and host demography is considered important in the differentiation and origin of P. knowlesi parasites and other closely related Plasmodium species, for which diversity is probably underestimated (16,28). We propose that the 2 distinct P. knowlesi populations currently circulating in Peninsular Malaysia and Malaysian of Borneo correspond to 2 independently evolving populations. The PkCOX1 haplotypes available for the P. knowlesi isolates from Peninsular Malaysia (Malayan, GenBank accession no. AB444106.1; Hackeri, accession no. AB444107.1; H, accession no. AB444108) fall within the Peninsular Malaysia cluster; corresponding PkA-type 18S rRNA sequences are not available.
The 3 P. knowlesi subspecies proposed to date (12) were found in distinct locations (Taiwan, Java, and Peninsular Malaysia). These parasites were not studied further and confirmation of their subspecific status remains to be confirmed, but no material suitable for genetic analysis is available. We note that evidence of potential hybrid forms was obtained from a single sample from a macaque from Sabah and for the H strain. The low frequency of such forms suggests a recent admixture through increased human movement between the 2 geographic regions of Malaysia, biologic factors that limit their fitness, or both. The clear geographic genetic differentiation and the indiscriminate distribution between human and macaque hosts for the parasites within each cluster strongly indicate that P. knowlesi became zoonotic independently in the 2 regions. Given that P. knowlesi has long been zoonotic, the data also indicate that human-to-human transmission has not been established over time.
While our study was underway, P. knowlesi populations from Sarawak were shown to be dimorphic across their genome (29,30). Furthermore, analyses based on microsatellite diversity identified 2 distinct parasite groups across the whole of Malaysia that strongly cluster with the macaque host species but not in the human samples: 1 was dominant in M. fascicularis macaques and the other in M. nemestrina macaques (31). These distinct groups do not correspond to the 2 distinct clusters we propose because their pattern of distribution is incongruent with the marked geographic distribution we report. It is likely that they represent a more recent population structure driven by adaptation to the macaque as vector and host. Considering the differences in the mutation rates between mitochondrial and nuclear genes and microsatellites (32,33), the data from the microsatellite analyses would be expected to detect recent population structuring, and our data from the nuclear and mitochondrial genes would reveal a more ancient geographic isolation.
In conclusion, it is clear that the P. knowlesi malaria parasites are not a homogeneous group but form a complex of related types, possibly including emerging subspecies, of which evolution and distribution has been shaped by past and recent events. It is equally clear that further analyses encompassing a larger number of genetic loci or whole genomes from sample sets collected in Malaysia, as well as in neighboring countries, will be needed to obtain a more comprehensive picture of the phylogeographic distribution and population structure of P. knowlesi. The zoonotic potential of these parasites constitutes a threat to the efforts to eliminate malaria in Southeast Asia. Thus, more epidemiologic and biologic investigations are necessary to help devise strategies that will minimize if not eliminate this threat.
Ms. Yusof is enrolled in the Master of Medical of Science program at Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia. Her research interests include evolutionary genetics of malaria parasites.
Acknowledgments
The authors thank Noradilah Marzuki, Marlindawati Mohd Ali, Hanapi Seli, Isma Lyna Ismail, Lin Bee Bee, Fatimah Haslina Abdullah, Azizah Mustafa, Kartina Md. Noor, Azizon Othman, Masturah Che Abdullah Hairolafizah Sebeki, and Sarimah Hashim for their assistance in collecting samples and in microscopic observation.
This study was funded by University of Malaya High Impact Research Fund (UM-MOHE UM.C/625/1/HIR/MOHE/MED/09) and Post-graduate Research Grant (PV044/2012A).
References
- World Health Organization. World malaria report 2014. Geneva, Switzerland: World Health Organization; 2014 [cited 2/19/2015 Feb 19]. http://www.who.int/malaria/publications/world_malaria_report_2014/en
- Knowles R, Das Gupta BM. A study of monkey-malaria, and its experimental transmission to man. (A preliminary report). Ind Med Gaz. 1932;67:301–20 [cited 2/19/2015 Feb 19]. http://www.cabdirect.org/abstracts/19322901628.html
- Sinton JA, Mulligan HW. A critical review of the literature relating to the identification of the malarial parasites recorded from monkeys of the families Cercopithecidæ and Colobidæ. Rec Mal Surv Ind. 1932;3:357–80 [cited 2/19/2015 Feb 19]. http://www.cabdirect.org/abstracts/19342900941.html
- Chin W, Contacos PG, Coatney GR, Kimball HR. A naturally acquired quotidian-type malaria in man transferable to monkeys. Science. 1965;149:865. DOIPubMedGoogle Scholar
- Singh B, Sung LK, Matusop A, Radhakrishnan A, Shamsul SSG, Cox-Singh J, A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–24. DOIPubMedGoogle Scholar
- Barber BE, William T, Dhararaj P, Anderios F, Grigg MJ, Yeo TW, Epidemiology of Plasmodium knowlesi malaria in north-east Sabah, Malaysia: family clusters and wide age distribution. Malar J. 2012;11:401. DOIPubMedGoogle Scholar
- Yusof R, Lau YL, Mahmud R, Fong MY, Jelip J, Ngian HU, High proportion of Knowlesi malaria in recent malaria cases in Malaysia. Malar J. 2014;13:168. DOIPubMedGoogle Scholar
- Cox-Singh J. Zoonotic malaria: Plasmodium knowlesi, an emerging pathogen. Curr Opin Infect Dis. 2012;25:530–6 .http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22710318&dopt=AbstractDOIPubMedGoogle Scholar
- Singh B, Daneshvar C. Human infections and detection of Plasmodium knowlesi. Clin Microbiol Rev. 2013;26:165–84. DOIPubMedGoogle Scholar
- Coatney GR, Collins WE, Warren M, Contacos PG. The primate malarias. Washington (DC): US Department of Health, Education and Welfare; 1971.
- Ahmed MA, Cox-Singh J. Plasmodium knowlesi—an emerging pathogen. ISBT Sci Ser. 2015;10(Suppl 1):134–40. DOIPubMedGoogle Scholar
- Garnham PCC. Malaria parasites and other haemosporidia. Oxford: Blackwell Scientific Publications; 1966.
- Pacheco MA, Battistuzzi FU, Junge RE, Cornejo OE, Williams CV, Landau I, Timing the origin of human malarias: the lemur puzzle. BMC Evol Biol. 2011;11:299. DOIPubMedGoogle Scholar
- Taylor JE, Pacheco MA, Bacon DJ, Beg MA, Machado RL, Fairhurst RM, The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: parasite genetic diversity in the Americas. Mol Biol Evol. 2013;30:2050–64. DOIPubMedGoogle Scholar
- Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Krettli AU, Early origin and recent expansion of Plasmodium falciparum. Science. 2003;300:318–2. DOIPubMedGoogle Scholar
- Muehlenbein MP, Pacheco MA, Taylor JE, Prall SP, Ambu L, Nathan S, Accelerated diversification of nonhuman primate malarias in Southeast Asia: adaptive radiation or geographic speciation? Mol Biol Evol. 2015;32:422–39. DOIPubMedGoogle Scholar
- Lee K-S, Divis PCS, Zakaria SK, Matusop A, Julin RA, Conway DJ, Plasmodium knowlesi: reservoir hosts and tracking the emergence in humans and macaques. PLoS Pathog. 2011;7:e1002015.DOIPubMedGoogle Scholar
- Singh B, Bobogare A, Cox-Singh J, Snounou G, Abdullah MS, Rahman HA. A genus- and species–specific nested polymerase chain reaction malaria detection assay for epidemiologic studies. Am J Trop Med Hyg. 1999;60:687–92.PubMedGoogle Scholar
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59.PubMedGoogle Scholar
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–20.DOIPubMedGoogle Scholar
- Earl DA, von Holdt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4:359–61 .DOIGoogle Scholar
- Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10:564–7.DOIPubMedGoogle Scholar
- Tajima F. The effect of change in population size on DNA polymorphism. Genetics. 1989;123:597–601.PubMedGoogle Scholar
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.PubMedGoogle Scholar
- Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133:693–709.PubMedGoogle Scholar
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.DOIPubMedGoogle Scholar
- Hanebuth T, Stattegger K, Grootes PM. Rapid flooding of the sunda shelf: A late-glacial sea-level record. Science. 2000;288:1033–5.DOIPubMedGoogle Scholar
- Pacheco MA, Reid MJC, Schillaci MA, Lowenberger CA, Galdikas BMF, Jones-Engel L, The origin of malarial parasites in orangutans. PLoS One. 2012;7:e34990.DOIPubMedGoogle Scholar
- Ahmed AM, Pinheiro MM, Divis PCS, Siner A, Zainudin R, Wong IT, Disease progression in Plasmodium knowlesi malaria is linked to variation in invasion gene family members. PLoS Negl Trop Dis. 2014;8:e3086.DOIPubMedGoogle Scholar
- Pinheiro MM, Ahmed MA, Millar SB, Sanderson T, Otto TD, Lu WC, Plasmodium knowlesi genome sequences from clinical isolates reveal extensive genomic dimorphism. PLoS One. 2015;10:e0121303.DOIPubMedGoogle Scholar
- Divis PC, Singh B, Anderios F, Hisam S, Matusop A, Kocken CH, Admixture in humans of two divergent Plasmodium knowlesi populations associated with different macaque host species. PLoS Pathog. 2015;11:e1004888. DOIPubMedGoogle Scholar
- Haasl RJ, Payseur BA. Multi-locus inference of population structure: a comparison between single nucleotide polymorphisms and microsatellites. Heredity. 2011;106:158–71. DOIPubMedGoogle Scholar
- Lehmann T, Blackston CR, Besansky NJ, Escalante AA, Collins FH, Hawley WA. The Rift Valley complex as a barrier to gene flow for Anopheles gambiae in Kenya: the mtDNA perspective. J Hered. 2000;91:165–8 .DOIPubMedGoogle Scholar
Figures
Tables
Cite This Article1These authors contributed equally to this article.
Table of Contents – Volume 22, Number 8—August 2016
| EID Search Options |
|---|
|
|
|
|
|
|
Please use the form below to submit correspondence to the authors or contact them at the following address:
Yee Ling Lau, Tropical Infectious Disease Research & Education Centre (TIDREC), Department of Parasitology, Faculty of Medicine, University of Malaya, 50603 Kuala Lumpur, Malaysia
Top